Multiple Mechanisms Suppress Signaling from the GPCR/cAMP/PKA Pathway
For cells to respond effectively to changes in their environment, they must not only activate a signaling pathway, but also down-modulate or terminate the response once it is no longer needed; otherwise, signal transduction pathways would remain “on” too long, or at too high a level, and the cell would become overstimulated.
Earlier we saw that multiple mechanisms can rapidly terminate the rhodopsin signal transduction pathway, including GAP proteins that stimulate the hydrolysis of GTP bound to Gαt, Ca2+-sensing proteins that activate guanylate cyclase, and phosphorylation of active rhodopsin by rhodopsin kinase followed by binding of arrestin (see Figure 15-21). In fact, most G protein–coupled receptors are modulated by multiple mechanisms that down-regulate their activity, as is exemplified by β-adrenergic receptors and others coupled to Gαs that activate adenylyl cyclase.
First, the intrinsic GTPase activity of Gαs converts the bound GTP to GDP, thus terminating its ability to activate its downstream target adenylyl cyclase. Importantly, the rate of hydrolysis of GTP bound to Gαs is enhanced when Gαs binds to adenylyl cyclase, lessening the duration of cAMP production; thus adenylyl cyclase functions as a GAP for Gαs·GTP. More generally, binding of most, if not all, Gα·GTP complexes to their respective effector proteins accelerates the rate of GTP hydrolysis.
Second, PDE acts to hydrolyze cAMP to 5′-AMP, terminating the cellular response. Thus the continuous presence of hormone at a high enough concentration is required for continuous activation of adenylyl cyclase and maintenance of an elevated cAMP level. Once the hormone concentration falls sufficiently, the cAMP level falls and all cellular responses quickly terminate.
Most GPCRs are also down-regulated by feedback repression, in which an end product of a signaling pathway blocks an early step in that pathway. For instance, when a Gαs protein–coupled receptor is exposed to hormonal stimulation for several hours, several serine and threonine residues in the cytosolic domain of the receptor become phosphorylated by PKA. The phosphorylated receptor can bind its ligand, but cannot efficiently activate Gαs; thus ligand bound to the phosphorylated receptor is less efficient in activating adenylyl cyclase then is ligand bound to the nonphosphorylated receptor. Because the activity of PKA is enhanced by the high cAMP level induced by any hormone that activates Gαs, prolonged exposure to one such hormone—say, epinephrine—desensitizes not only β-adrenergic receptors, but also other Gαs protein–coupled receptors that are phosphorylated by PKA, even though they bind different ligands (e.g., glucagon receptors in the liver). This cross-regulation is called heterologous desensitization.
Several residues in the cytosolic domain of the β-adrenergic receptor, different from those phosphorylated by PKA, are phosphorylated by the enzyme β-adrenergic receptor kinase (BARK), but only when epinephrine or an agonist is bound to the receptor and thus the receptor is in its active conformation. BARK is a member of the same kinase family as rhodopsin kinase, and its action is similar to the phosphorylation and down-modulation of activated rhodopsin by rhodopsin kinase (see Figure 15-21). This process is termed homologous desensitization because only those receptors that are in their active conformations are subject to deactivation by phosphorylation.
We noted that binding of arrestin to extensively phosphorylated opsin completely inhibits activation of coupled G proteins by activated opsin (see Figure 15-21). A related protein, termed β-arrestin, plays a similar role in silencing other G protein–coupled receptors, including β-adrenergic receptors (Figure 15-32).
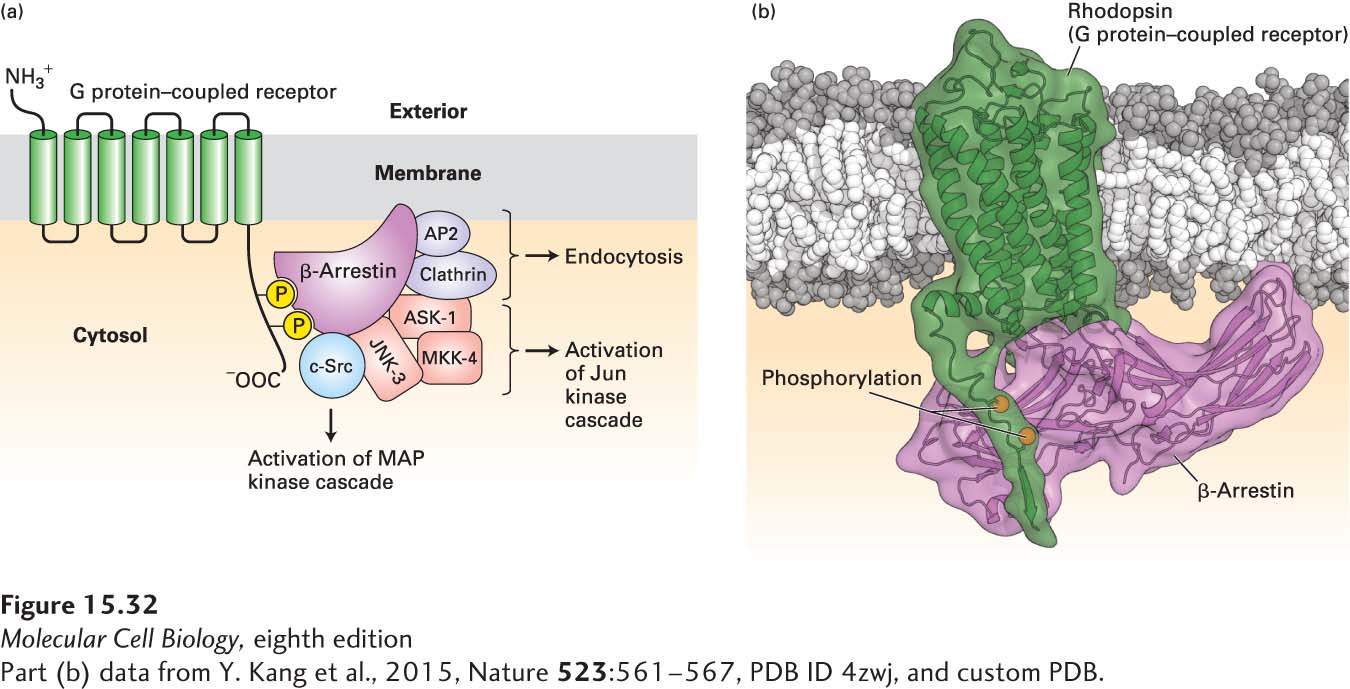
FIGURE 15-32 Binding of β-arrestin to phosphorylated GPCRs triggers receptor desensitization and activation of several different signal transduction proteins. (a) β-Arrestin binds to specific phosphorylated serine and threonine residues in the C-terminal segment of G protein–coupled receptors (GPCRs). Clathrin and AP2, two other proteins bound by β-arrestin, promote endocytosis of the receptor (see Figure 14-29). β-Arrestin also functions in transducing signals from activated receptors by binding to and activating several cytosolic protein kinases. Src activates the MAP kinase pathway, leading to phosphorylation of key transcription factors (see Chapter 16). Interaction of β-arrestin with three other proteins, including JNK-3 (a Jun N-terminal kinase), results in phosphorylation and activation of another transcription factor, Jun. See W. Miller and R. J. Lefkowitz, 2001, Curr. Opin. Cell Biol. 13:139, and K. Pierce et al., 2002, Nat. Rev. Mol. Cell Biol. 3:639. (b) Three-dimensional structure of rhodopsin bound to arrestin. Arrestin binds to segments of the C-terminal cytosolic alpha helix of activated rhodopsin that includes the two phosphorylated resides as well as to parts of transmembrane helix 7.
[Part (b) data from Y. Kang et al., 2015, Nature 523:561-567, PDB ID 4zwj, and custom PDB.]
An additional function of β-arrestin in regulating cell-surface receptors was initially suggested by the observation that disappearance of β-adrenergic receptors from the cell surface in response to ligand binding is stimulated by overexpression of BARK and β-arrestin. Subsequent studies revealed that β-arrestin binds not only to phosphorylated GPCRs, but also to clathrin and an associated protein termed AP2, two key components of the coated vesicles that are involved in endocytosis from the plasma membrane (see Figure 15-32; see also Chapter 14). These interactions promote the formation of coated pits and endocytosis of the associated receptors, thereby decreasing the number of receptors exposed on the cell surface. Eventually some of the internalized receptors are degraded intracellularly, and some are de-phosphorylated in endosomes. Following dissociation of β-arrestin, the resensitized (de-phosphorylated) receptors are recycled to the cell surface in a manner similar to the recycling of the LDL receptor (see Chapter 14).
In addition to its role in regulating receptor activity, β-arrestin functions as an adapter protein in transducing signals from G protein–coupled receptors to the nucleus (see Chapter 16). The GPCR-arrestin complex acts as a scaffold for the binding and activation of several cytosolic kinases (see Figure 15-32), which we discuss in detail in subsequent chapters. These kinases include Src, a cytosolic protein tyrosine kinase that activates the MAP kinase pathway and other pathways leading to the transcription of genes needed for cell division (see Chapters 16 and 19). A complex of three arrestin-bound proteins, including a Jun N-terminal kinase (JNK-3), initiates a kinase cascade that ultimately activates the Jun transcription factor, which promotes expression of certain growth-promoting enzymes and other proteins that help cells respond to stresses. Thus the BARK–β-arrestin pathway, originally just thought to suppress signaling by GPCRs, actually functions as a switch, turning off signaling by G proteins and turning on other signaling pathways. The multiple functions of β-arrestin illustrate the importance of adapter proteins in both regulating signaling and transducing signals from cell-surface receptors.