The DNA Damage Response System Halts Cell Cycle Progression When DNA Is Compromised
The complete and accurate duplication of the genetic material is essential for cell division. If cells enter mitosis when DNA is incompletely replicated or otherwise damaged, genetic changes occur. In many instances, those changes will lead to cell death, but as we will see in Chapter 24, they can also lead to genetic alterations that result in loss of control over cell growth and division and, eventually, cancer. This risk is underscored by the finding that many proteins involved in sensing DNA damage and its repair are frequently found mutated in human cancers.
The enzymes that replicate DNA are highly accurate, but their exactness is not enough to ensure complete accuracy during DNA synthesis. Furthermore, environmental insults such as x-rays and UV light can cause DNA damage, and this damage must be repaired before a cell’s entry into mitosis. Cells have a DNA damage response system in place that senses many different types of DNA damage and responds by activating repair pathways and halting cell cycle progression until the damage has been repaired. Cell cycle arrest can occur in G1, S phase, or G2, depending on whether DNA damage occurred before cell cycle entry or during DNA replication. In multicellular organisms, the strategy to deal with particularly severe DNA damage is different. Rather than attempting to repair the damage, cells undergo programmed cell death or apoptosis, a mechanism that we will discuss in detail in Chapter 21.
DNA damage exists in many different forms and degrees of severity. A break of the DNA helix, known as a double-strand break, is perhaps the most severe form of damage because such a lesion would almost certainly lead to DNA loss if mitosis ensued in its presence. More subtle defects include single-strand breaks, structural changes in nucleotides, and DNA mismatches. For our discussion here, it is important to note that cells have sensors for all these different types of damage. These sensors scan the genome and, when they detect a lesion, assemble signaling and repair factors at the site of the lesion.
Central to the detection of these different types of lesions is a pair of homologous protein kinases called ATM (for ataxia telangiectasia mutated) and ATR (for ataxia telangiectasia and Rad3-related protein). These proteins were identified and characterized because mutations in their encoding genes cause ataxia telangiectasia and Seckel syndrome, respectively. Both protein kinases are recruited to sites of DNA damage. They then initiate the sequential recruitment of adapter proteins and another set of protein kinases called Chk1 and Chk2. Those kinases then activate repair mechanisms and cause cell cycle arrest or apoptosis in animals (Figure 19-29). ATR and ATM recognize different types of DNA damage. ATM is very specialized in that it responds only to double-strand breaks. ATR is able to recognize more diverse types of DNA damage, such as stalled replication forks, DNA mismatches, damaged nucleotides, and double-strand breaks. ATR recognizes these diverse types of damage because all of them contain some amount of single-stranded DNA, either as part of the damage itself or because repair enzymes create single-stranded DNA as part of the repair process. The association of ATR with single-stranded DNA is thought to activate its protein kinase activity, leading to the recruitment of adapter proteins whose function is to recruit and help activate the Chk1 kinase. Active Chk1 then induces repair pathways and inhibits cell cycle progression.
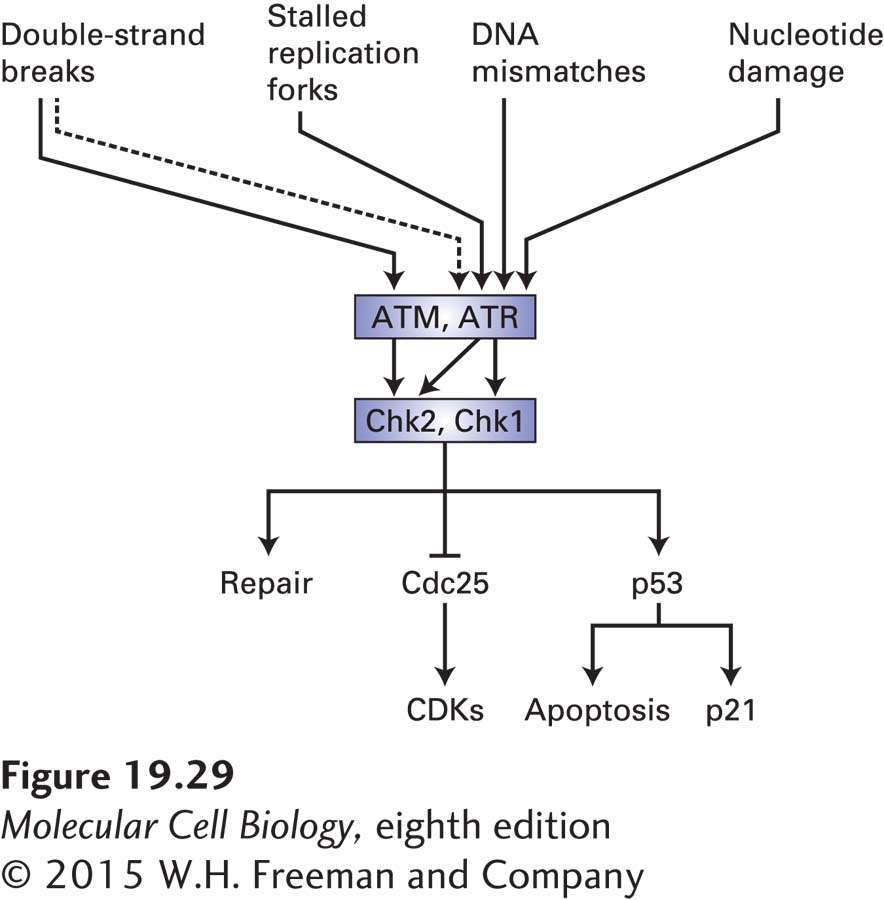
FIGURE 19-29 The DNA damage response system. The protein kinases ATM and ATR are activated by damaged DNA. ATR responds to a variety of DNA damage—most likely to the single-stranded DNA that exists either as a result of the damage itself or as a result of repair. ATM is specifically activated by double-strand breaks. Because double-strand breaks are converted into single-stranded DNA as a part of the repair process, they also, albeit indirectly (as depicted by a dashed line), activate ATR. ATM and ATR, once activated by DNA damage, activate another pair of related protein kinases, Chk1 and Chk2. These kinases then induce the DNA repair machinery and cause cell cycle arrest by inhibiting Cdc25. In metazoan cells, Chk1 and Chk2 also activate the transcription factor p53, which induces cell cycle arrest by inducing transcription of the CKI p21. When the DNA damage is severe, p53 induces apoptosis.
Chk1 and Chk2 halt the cell cycle. These protein kinases inhibit Cdc25 by phosphorylating that phosphatase on sites that are distinct from the CDK-activating phosphorylation sites (see Figure 19-29). When the DNA damage occurs during G1, Cdc25A inhibition results in inhibition of G1/S phase CDKs and S phase CDKs (Figure 19-30; see also Figure 19-18). As a result, these kinases cannot initiate DNA replication. When the DNA damage occurs during S phase or in G2, Cdc25C inhibition by Chk1/2 results in the inhibition of mitotic CDKs and hence arrest in G2. Active DNA replication also inhibits entry into mitosis. ATR continues to inhibit Cdc25C via Chk1 until all replication forks complete DNA replication and disassemble. This mechanism makes the initiation of mitosis dependent on the completion of chromosome replication. Finally, cells also sense DNA replication stress that results in stalling or slowing of replication forks. Such stress triggers activation of the ATR-Chk1 checkpoint pathway and results in down-regulation of S phase CDK activity and prevents the firing of late-replicating origins.
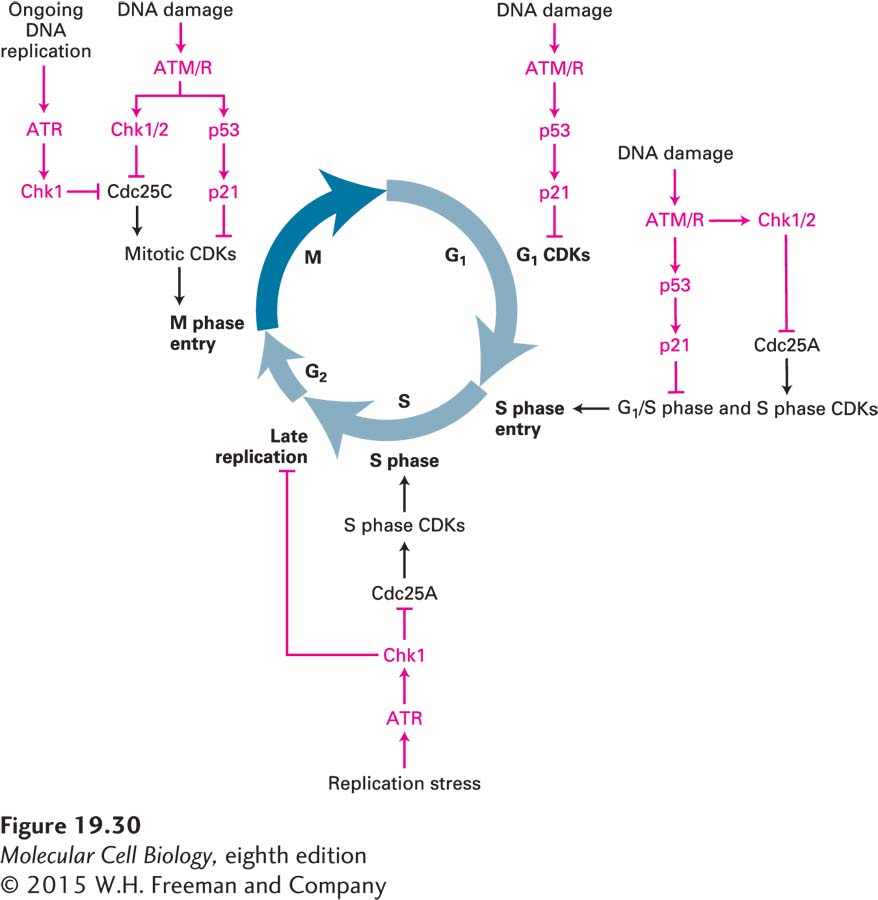
FIGURE 19-30 Overview of DNA damage checkpoint controls in the cell cycle. During G1, the p53-p21 pathway inhibits G1 CDKs. During ongoing DNA replication and in response to replication stress (slow DNA replication fork movement or DNA replication fork collapse), the ATR-Chk1 protein kinase cascade phosphorylates and inactivates Cdc25C, thereby preventing the activation of mitotic CDKs and inhibiting entry into mitosis. In response to DNA damage, the ATM or ATR protein kinases (ATM/R) inhibit Cdc25 via the Chk1/2 protein kinases. They also activate p53, which induces production of the CKI p21. During G1, the DNA damage checkpoint pathway inhibits Cdc25A, inhibiting G1/S phase CDKs and S phase CDKs, and thereby blocking entry into or passage through S phase. During G2, ATM/R and Chk1/2 inhibit Cdc25C. The p53-p21 pathway is also activated. Red symbols indicate pathways that inhibit progression through the cell cycle.
Chk1-mediated inhibition of the Cdc25 family of phosphatases is not the only mechanism whereby DNA damage or incomplete replication inhibits cell cycle progression. As we will see below, DNA damage leads to the activation of p53, a transcription factor that induces the expression of the gene encoding the CDK inhibitor p21. The p21 binds to and inhibits all metazoan cyclin-CDK complexes. As a result, cells are arrested in G1 and G2 (see Figure 19-30).
As mentioned above, ATM recognizes only double-strand breaks (see Figure 19-29). This protein kinase is directly recruited to the DNA ends by a complex known as the MRN complex, which binds to the broken ends and holds them together. Activated ATM then phosphorylates and activates Chk2 and recruits repair proteins. These repair proteins initiate homologous recombination, as described in Chapter 5. This process involves the creation of single-stranded overhangs, which in turn recruit and activate ATR and its effectors, further enhancing the DNA damage response. ATM can also recruit an alternative repair pathway by which two double-strand breaks are directly fused with each other in a repair process known as nonhomologous end joining. Like ATR, activated ATM also halts cell cycle progression by Chk2-mediated inhibition of Cdc25, thus preventing activation of CDKs. This inhibition can occur in G1 or G2.
A key effector of the DNA damage response in metazoan cells is the transcription factor p53 (see Figure 19-29). It is known as a tumor suppressor because its normal function is to limit cell proliferation in the face of DNA damage. This protein is extremely unstable and generally does not accumulate to high enough levels to stimulate transcription under normal conditions. The instability of p53 results from its ubiquitinylation by a ubiquitin-protein ligase called Mdm2 and its subsequent proteasomal degradation. Rapid degradation of p53 is inhibited by ATM and ATR, which phosphorylate p53 at a site that interferes with Mdm2 binding. This and other modifications of p53 in response to DNA damage greatly increase its ability to activate transcription of specific genes that help the cell cope with DNA damage. One of these genes encodes the CKI p21 (see Figure 19-29).
Under some circumstances, such as when DNA damage is extensive, p53 also activates expression of genes that lead to apoptosis, the process of programmed cell death that normally occurs in specific cells during the development of multicellular animals. In metazoans, the p53 response evolved to induce apoptosis (see Chapter 21) in the face of extensive DNA damage, presumably to prevent the accumulation of multiple mutations that might convert a normal cell into a cancer cell. The dual role of p53 in both cell cycle arrest and the induction of apoptosis may account for the observation that nearly all cancer cells have mutations in both alleles of the p53 gene or in the pathways that stabilize p53 in response to DNA damage (see Chapter 24). The consequences of mutations in p53, ATM, and Chk2 provide dramatic examples of the significance of cell cycle checkpoint pathways to the health of a multicellular organism.