The Amino Acid Sequence of a Protein Determines How It Will Fold
While the constraints of backbone bond angles seem very restrictive, any polypeptide chain containing only a few residues could, in principle, still fold into many conformations. For example, if the Φ and Ψ angles were limited to only eight combinations, an n-residue-long peptide would potentially have 8n conformations; for even a small polypeptide of only 10 residues, that’s about 8.6 million possible conformations! In general, however, the native state of any particular protein that is not intrinsically disordered adopts only one or a few very closely related conformations; for the vast majority of these proteins, the native state is a stably folded form of the molecule and the one that permits it to function normally. In thermodynamic terms, the native state is usually the conformation with the lowest free energy (G) (see Chapter 2).
What features of natively well-ordered proteins limit their folding from so many potential conformations to just one or a few? The properties of the side chains (e.g., size, hydrophobicity, ability to form hydrogen and ionic bonds), together with their particular sequence along the polypeptide backbone, impose key restrictions. For example, a large side chain, such as that of tryptophan, might sterically block one region of the chain from packing closely against another region, whereas a side chain with a positive charge, such as that of arginine, might attract a segment of the polypeptide that has a complementary negatively charged side chain (e.g., aspartic acid). Another example we have already discussed is the effect of the aliphatic side chains in heptad repeats in promoting the association of helices and the consequent formation of coiled coils. Thus a polypeptide’s primary structure determines its secondary, tertiary, and quaternary structures.
The initial evidence that the information necessary for a protein to fold properly is encoded in its amino acid sequence came from in vitro studies (in test tubes) on the refolding of purified proteins, especially the Nobel Prize–winning studies in the 1960s by Christian Anfinsen of the refolding of ribonuclease A, an enzyme that cleaves RNA. Others had previously shown that various chemical and physical perturbations can disrupt the weak noncovalent interactions that stabilize the native conformation of a protein, leading to the loss of its normal tertiary structure. The disruption of a protein’s structure (and this can include secondary as well as tertiary structure) is called denaturation. Denaturation can be induced by thermal energy from heat, extremes of pH that alter the charges on amino acid side chains, or exposure to denaturants such as urea or guanidine hydrochloride at concentrations of 6–8 M, all of which disrupt structure-stabilizing noncovalent interactions. Treatment with reducing agents, such as β-mercaptoethanol, that break disulfide bonds can further destabilize disulfide-containing proteins. Under denaturing conditions, a population of uniformly folded protein molecules is destabilized and converted into a collection of many unfolded, or denatured, molecules that have many different non-native and biologically inactive conformations. As we have seen, a large number of possible non-native conformations exist (e.g., 8n − 1). There are two broad classes of non-native conformations seen in proteins: (1) monomeric unfolded or denatured structures and (2) aggregates, which can either be amorphous or have a well-organized structure, as is the case for the disease-associated amyloid fibrils described later in this chapter. In principle, aggregates can comprise many copies of a single protein (homogeneous aggregates) or contain a mixture of distinct proteins (heterogeneous aggregates).
The spontaneous unfolding of proteins under denaturing conditions is not surprising, given the substantial increase in entropy that occurs because a denatured protein can adopt many non-native conformations (increased disorder). What is striking, however, is that when a pure sample of a single type of unfolded protein in a test tube is shifted back very carefully to normal conditions (body temperature, normal pH levels, reduction in the concentration of denaturants), some denatured polypeptides can spontaneously refold into their native, biologically active states, as in Anfinsen’s experiments. This kind of refolding experiment, as well as studies showing that synthetic proteins made chemically can fold properly, established that the information contained in a protein’s primary structure can be sufficient to direct correct refolding. Newly synthesized proteins appear to fold into their proper conformations just as denatured proteins do. The observed similarity in the folded, three-dimensional structures of proteins with similar amino acid sequences, noted in Section 3.1, provided additional evidence that the primary sequence also determines protein folding in vivo (in live organisms). It appears that formation of secondary structures and structural motifs occurs early in the folding process, followed by assembly of more complex structural domains, which then associate into more complex tertiary and quaternary structures (Figure 3-16).
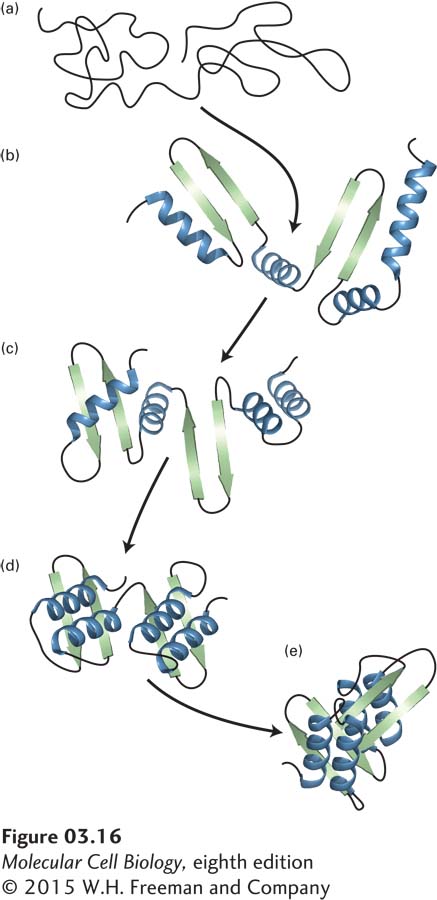
FIGURE 3-16 Hypothetical protein-folding pathway. Folding of a monomeric protein follows the structural hierarchy of primary (a) → secondary (b–d) → tertiary (e) structure. Formation of small structural motifs (c) appears to precede formation of domains (d) and the final tertiary structure (e).