Radioisotopes Are Indispensable Tools for Detecting Biological Molecules
A sensitive method for tracking a protein or other biological molecule is by detecting the radioactivity emitted from a radiolabel introduced into the molecule. In a radiolabeled molecule, at least one atom is present in a radioactive form, called a radioisotope.
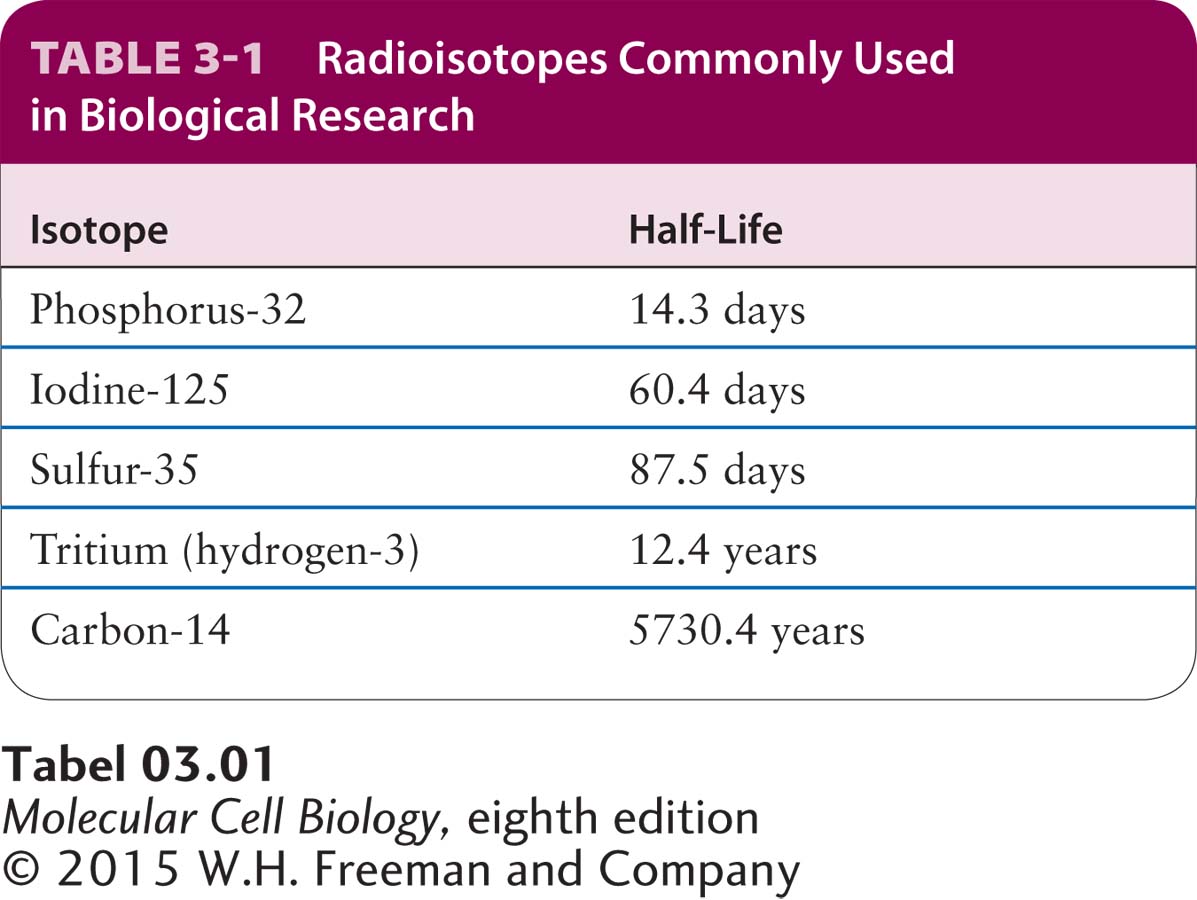
Radioisotopes Useful in Biological Research Hundreds of biological molecules (e.g., amino acids, nucleosides, and numerous other small-molecule metabolites) labeled with various radioisotopes are commercially available. These preparations vary considerably in their specific activity, which is the amount of radioactivity per unit of material, measured in disintegrations per minute (dpm) per millimole (mmol). The specific activity of a labeled compound depends on the radioisotope’s half-life, the time required for half the atoms to undergo radioactive decay, which releases the detectable radiation. In general, the shorter the half-life of a radioisotope, the higher its specific activity (Table 3-1). The specific activity of a labeled compound must be high enough for accurate detection of its emitted radiation.
A common approach to radiolabeling macromolecules (proteins, RNA, DNA) in cells is to add a radiolabeled biosynthetic precursor to the extracellular medium [e.g., 3H- or 35S-labeled amino acids, 32P-labeled phosphate (precursor for 32P-labeled ATP), or 3H-labeled nucleic acid precursors such as deoxythymidine (also simply called thymidine) or 32P-labeled phosphate]. The precursor enters the cells via transporters (see Chapter 11) and is incorporated into newly synthesized macromolecules by the cells (see Chapter 5). For example, methionine and cysteine labeled with sulfur-35 (35S) are widely used to label cellular proteins because preparations of these amino acids with high specific activities (>1015 dpm/mmol) are available. Kinases within cells (or used in vitro) can transfer a 32P-labeled phosphate from 32P-labeled ATP to label phosphoproteins. Likewise, commercial preparations of 3H-labeled nucleic acid precursors have much higher specific activities than those of the corresponding 14C-labeled preparations. In most experiments, the former are preferable because they allow RNA or DNA to be adequately labeled a shorter time after incorporation or require a smaller cell sample. Various phosphate-containing compounds in which the phosphorus atom is the radioisotope phosphorus-32 are readily available. Because of their high specific activity, 32P-labeled nucleotides are routinely used to label nucleic acids in cell-free systems.
Labeled compounds in which a radioisotope replaces atoms normally present in the molecule have virtually the same chemical properties as the corresponding unlabeled compounds. Enzymes, for instance, generally cannot distinguish between substrates labeled in this way and their unlabeled substrates. The presence of such radioactive atoms is indicated with the isotope in brackets (no hyphen) as a prefix (e.g., [3H]leucine). In contrast, labeling of almost any biomolecule (e.g., protein or nucleic acid) with the radioisotope iodine-125 (125I) requires the covalent addition of 125I to a molecule that normally does not have iodine as part of its structure. Because this labeling procedure modifies the chemical structure, the biological activity of the labeled molecule may differ somewhat from that of the unlabeled form. The presence of such radioactive atoms is indicated with the isotope as a prefix followed a hyphen (no bracket) (e.g., 125I-trypsin). Standard methods for labeling proteins with 125I result in covalent attachment of the 125I primarily to the aromatic rings of tyrosine side chains (mono- and diiodotyrosine). Nonradioactive isotopes are finding increasing use in cell biology, especially in nuclear magnetic resonance studies and in mass spectroscopy applications, as will be explained below.
Labeling Experiments and Detection of Radiolabeled Molecules Whether labeled compounds are detected by autoradiography—exposure of the sample on a two-dimensional detector (photographic emulsion or electronic detector)—or their radioactivity is measured in an appropriate “counter,” the amount of a radiolabeled compound in a sample can be determined with great precision.
In one use of autoradiography, a tissue, cell, or cell constituent is labeled with a radioactive molecule, unassociated radioactive material is washed away, and the structure of the sample is stabilized either by chemically cross-linking the macromolecules in the sample (“fixation”) or by freezing it. The sample is then overlaid with a photographic emulsion that is sensitive to radiation. Development of the emulsion yields small silver grains whose distribution corresponds to that of the radioactive material and is usually detected by microscopy. Autoradiographic studies of whole cells were crucial in determining the intracellular sites where various macromolecules are synthesized and the subsequent movements of those macromolecules within cells. Various techniques employing fluorescence microscopy, which we describe in Chapter 4, have largely supplanted autoradiography for studies of this type. However, autoradiography is sometimes used in various assays for detecting specific isolated DNA or RNA sequences at specific tissue locations (see Chapter 6) in a technique referred to as in situ hybridization.
Quantitative measurements of the amount of radioactivity in a labeled material are performed with several different instruments. A Geiger counter measures ions produced in a gas by the β particles or γ rays emitted from a radioisotope. These instruments are mostly handheld devices used to monitor radioactivity in the laboratory to protect investigators from excess exposure. In a scintillation counter, a radiolabeled sample is mixed with a liquid containing a fluorescent compound that emits a flash of light when it absorbs the energy of the β particles or γ rays released in the decay of the radioisotope; a phototube in the instrument detects and counts these light flashes. Phosphorimagers detect radioactivity using a two-dimensional array detector, storing digital data on the number of disintegrations per minute per small pixel of surface area. These instruments, which can be thought of as a kind of reusable electronic film, are commonly used to quantify radioactive molecules separated by gel electrophoresis and are replacing photographic emulsions for this purpose.
Combinations of labeling and biochemical techniques and of visual and quantitative detection methods are often employed in labeling experiments. For instance, to identify the major proteins synthesized by a particular cell type, a sample of the cells is incubated with a radiolabeled amino acid (e.g., [35S]methionine) for a few minutes, during which time the labeled amino acid enters the cells and mixes with the cellular pool of unlabeled amino acids, and some of it is biosynthetically incorporated into newly synthesized proteins. Subsequently, unincorporated radiolabeled amino acid is washed away from the cells. The cells are harvested, and the mixture of cellular proteins is extracted from the cells (for example, by a detergent solution) and then separated by any of the methods commonly used to resolve complex protein mixtures into individual components. Gel electrophoresis in combination with autoradiography or phosphorimager analysis is often the method of choice. The radioactive bands in the gel correspond to newly synthesized proteins, which have incorporated the radiolabeled amino acid. To detect a specific protein of interest, rather than the entire ensemble of biosynthetically radiolabeled proteins, a specific protein can be isolated by immunoprecipitation. The precipitate is then solubilized, for example, in an SDS-containing buffer, and the sample is analyzed by SDS-PAGE followed by autoradiography to detect the protein that is radioactively labeled. In this type of experiment, a fluorescent compound that is activated by the radiation (“scintillator”) may be infused into the gel on completion of the electrophoretic separation so that the light emitted can be used to detect the presence of the labeled protein, using either film or a two-dimensional electronic detector. An example is shown in the experiment described in Figure 3-42. This method is particularly useful for weak β emitters such as 3H.
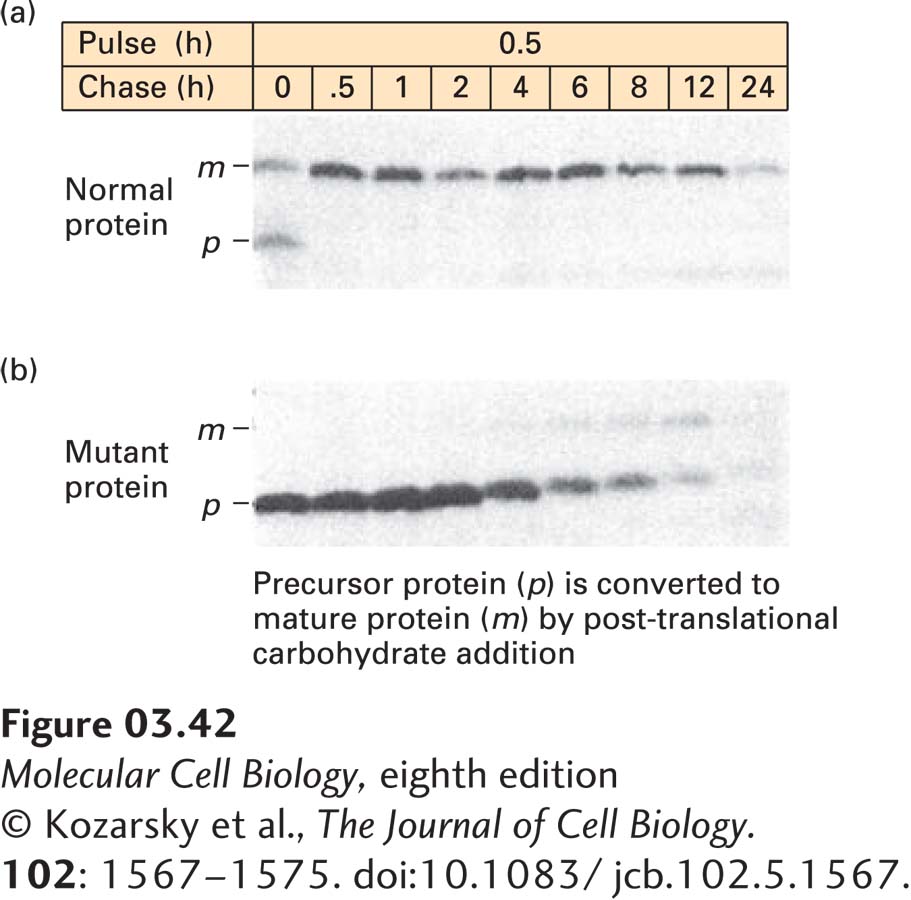
EXPERIMENTAL FIGURE 3-42 Pulse-chase experiments can track the pathway of protein modification within cells. (a) To follow the fate of a specific newly synthesized protein in cells, cells were incubated with [35S]methionine for 0.5 hours (the pulse) to label all newly synthesized proteins, and any radioactive amino acid not incorporated into the cells was then washed away. The cells were further incubated (the chase) for varying times up to 24 hours, and samples from each time of chase were subjected to immunoprecipitation to isolate one specific protein (here the low-density lipoprotein receptor). SDS-PAGE of the immunoprecipitates followed by autoradiography permitted visualization of the target protein, which is initially synthesized as a small precursor (p) and then rapidly modified to a larger mature form (m) by addition of carbohydrates. About half of the labeled protein was converted from p to m during the pulse; the rest was converted after 0.5 hours of chase. The protein remained stable for 6–8 hours before it began to be degraded (as indicated by reduced band intensity). (b) The same experiment was performed in cells in which a mutant form of the protein is made. The mutant p form cannot be properly converted to the m form, and it is more quickly degraded than the normal protein.
[© Kozarsky et al., The Journal of Cell Biology. 102: 1567–1575. doi:10.1083/jcb.102.5.1567.]
Pulse-chase experiments are particularly useful for tracing changes in the intracellular location of proteins or the modification of a protein or metabolite over time. In this experimental protocol, a cell sample is exposed to a radiolabeled compound that can be incorporated into or otherwise attached to a cellular molecule of interest—the “pulse”—for a brief period. The pulse ends when the unincorporated radiolabeled molecules are washed away and the cells are exposed to a vast excess of the identical, but unlabeled, compound to dilute the radioactivity of any remaining, but unincorporated, radiolabeled compound. This procedure prevents any incorporation of significant amounts of radiolabel after the “pulse” period and initiates the “chase” period (see Figure 3-42). Samples taken periodically during the chase period are assayed to determine the location or chemical form of the radiolabel as a function of time. Pulse-chase experiments in which the radiolabeled protein is detected by autoradiography after immunoprecipitation and SDS-PAGE are often used to follow the rate of synthesis, modification, and degradation of proteins. In these experiments, radiolabeled amino acid precursors are added during the pulse, and the amounts and characteristics of the radiolabeled target protein are detected during the chase. One can thus observe postsynthetic modifications of the protein, such as the covalent addition of sugars (see Chapters 13 and 14) or proteolytic cleavage, that change its electrophoretic mobility, as well as the rate of degradation of the protein, which is detected as the loss of signal with increasing time of chase. A classic use of the pulse-chase technique with autoradiography was in studies that elucidated the pathway traversed by secreted proteins from their site of synthesis in the endoplasmic reticulum to the cell surface (see Chapter 14).