Mass Spectrometry Can Determine the Mass and Sequence of Proteins
Mass spectrometry (MS) is a powerful technique for characterizing proteins, especially for determining the mass of a protein or fragments of a protein. With such information in hand, it is also possible to determine part or all of the protein’s sequence. This method permits the accurate direct determination of the ratio of the mass (m) of a charged molecule (molecular ion) to its charge (z), or m/z. Additional techniques are then used to deduce the absolute mass of the molecular ion.
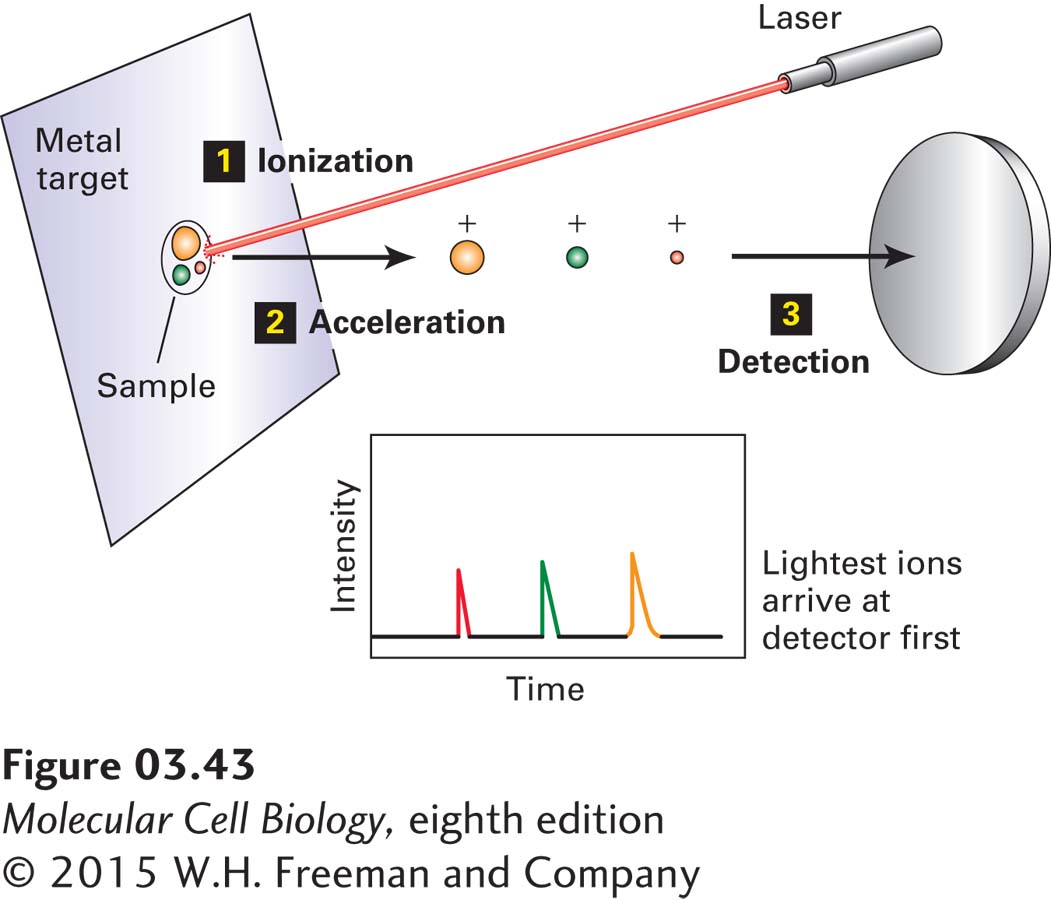
EXPERIMENTAL FIGURE 3-43 Molecular mass can be determined by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. In a MALDI-TOF mass spectrometer, pulses of light from a laser ionize a protein or peptide mixture that is absorbed on a metal target (step 1). An electric field in the mass analyzer accelerates the ions in the sample toward the detector (steps 2 and 3). The time it takes an ion to reach the detector is proportional to the square root of the mass-to-charge (m/z) ratio. Among ions having the same charge, the smaller ions move faster (shorter time to the detector). The molecular weight of each ion from the sample is calculated using the time of flight of a standard.
All mass spectrometers have four key features. The first is an ion source, from which charge, usually in the form of protons, is transferred to the peptide or protein molecules under study (ionization). Their conversion to ions occurs in the presence of a high electric field, which then directs the charged molecular ions into the second key component, the mass analyzer. The mass analyzer, which is always in a high vacuum chamber, physically separates the ions on the basis of their differing mass-to-charge (m/z) ratios. The separated ions are subsequently directed to strike a detector, the third key component, which provides a measure of the relative abundances of each of the ions in the sample. The fourth essential component is a computerized data system that is used to calibrate the instrument; to acquire, store, and process the resulting data; and often to direct the instrument to automatically collect additional specific types of data from the sample, based on the initial observations. This type of automated feedback is used for the tandem MS (MS/MS) peptide-sequencing methods described below.
The two most frequently used methods of generating ions of proteins and protein fragments are (1) matrix-assisted laser desorption/ionization (MALDI) and (2) electrospray (ES). In MALDI (Figure 3-43), the peptide or protein sample is mixed with a low-molecular-weight, UV-absorbing organic acid (the matrix) and then dried on a metal target. Energy from a laser ionizes and vaporizes the sample, producing singly charged molecular ions from the constituent molecules. In ES (Figure 3-44a), a sample of peptides or proteins in solution is converted into a fine mist of tiny droplets by spraying through a narrow capillary at atmospheric pressure. The droplets are formed in the presence of a high electric field, which renders them highly charged. The solvent evaporates from the droplets in their short flight (mm) to the entrance of the mass spectrometer’s mass analyzer, forming multiply charged ions from the peptides and proteins. The gaseous ions are transferred into the mass analyzer region of the MS, where they are then accelerated by electric fields and separated by the mass analyzer on the basis of their m/z.
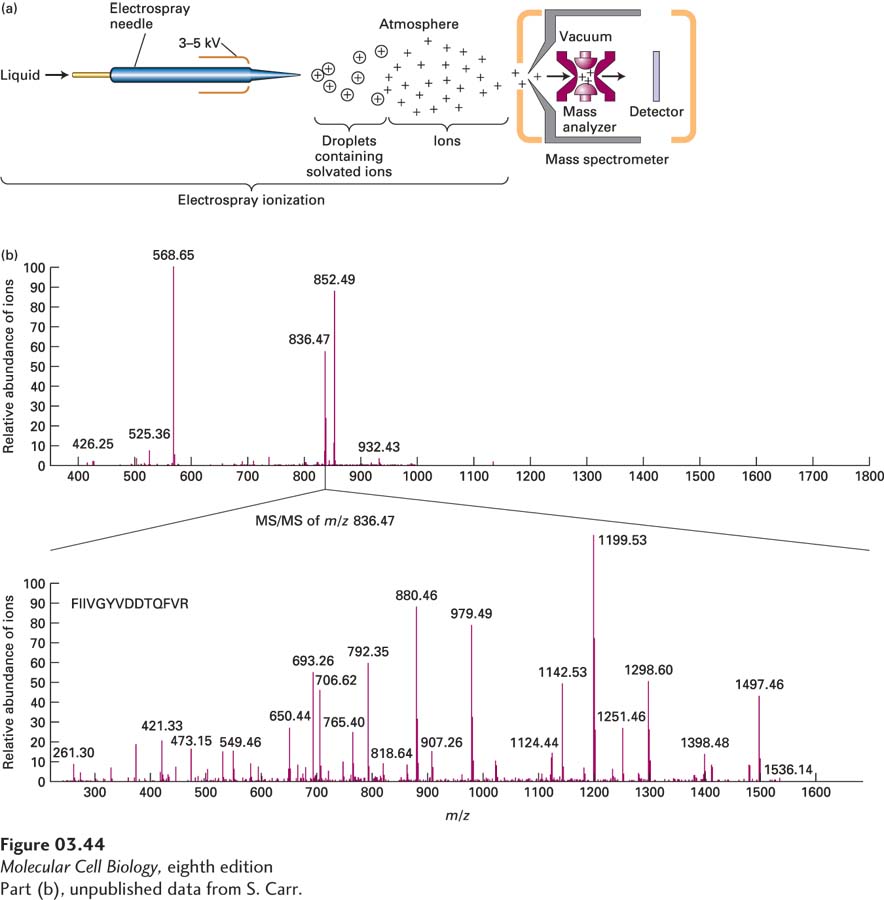
EXPERIMENTAL FIGURE 3-44 Molecular mass of proteins and peptides can be determined by electrospray ionization ion-trap mass spectrometry. (a) Electrospray (ES) ionization converts proteins and peptides in a solution into highly charged gaseous ions by passing the solution through a needle (forming the droplets) that has a high voltage across it (charging the droplets). Evaporation of the solvent produces gaseous ions that enter a mass spectrometer. The ions are analyzed by an ion-trap mass analyzer that then directs ions to the detector. (b) Top panel: Mass spectrum of a mixture of three major and several minor peptides from the mouse H-2 class I histocompatibility antigen Q10 α chain is presented as the relative abundance of the ions striking the detector (y axis) as a function of the mass-to-charge (m/z) ratio (x axis). Bottom panel: In an MS/MS instrument such as the ion trap shown in part (a), a specific peptide ion can be selected for fragmentation into smaller ions that are then analyzed and detected. The MS/MS spectrum (also called the product-ion spectrum) provides detailed structural information about the parent ion, including sequence information for peptides. Here the ion with an m/z of 836.47 was selected and fragmented and the m/z mass spectrum of the product ions measured. Note there is no longer an ion with an m/z of 836.47 present because it was fragmented. From the varying sizes of the product ions, the understanding that peptide bonds are often broken in such experiments, the known m/z values for individual amino acid fragments, and database information, the sequence of the peptide, FIIVGYVDDTQFVR, can be deduced.
[Part (b), unpublished data from S. Carr.]
The two most frequently used types of mass analyzers are time-of-flight (TOF) instruments and ion traps. TOF instruments exploit the fact that the time it takes an ion to pass through the length of the mass analyzer before reaching the detector is proportional to the square root of m/z (smaller ions move faster than larger ones with the same charge; see Figure 3-43). In ion-trap analyzers, tunable electric fields are used to capture, or “trap,” ions with a specific m/z and to sequentially pass the trapped ions out of the mass analyzer onto the detector (see Figure 3-44a). By varying the electric fields, researchers can examine ions with a wide range of m/z values one by one, producing a mass spectrum, which is a graph of m/z (x axis) versus relative abundance, determined by the intensity of the signal measured by the detector (y axis) (Figure 3-44b, top panel).
In tandem, or MS/MS, instruments, any given parent ion in the original mass spectrum (see Figure 3-44b, top panel) can be chosen (mass-selected) for further analysis. The chosen ions are transferred into a second chamber in which they are broken into smaller fragment ions by collision with an inert gas, and then the m/z and relative abundances of the resulting fragment ions are measured in a second MS analyzer (Figure 3-44b, bottom panel, see also Figure 3-47 later in this chapter). These multiple mass analysis and fragmentation steps all take place within the same machine in about 0.1 seconds per selected parent ion. The fragmentation and subsequent mass analysis permit the sequences of short peptides (<25 amino acids) to be determined because collisional fragmentation occurs primarily at peptide bonds, so the differences in masses between the multiple ion fragments generated correspond to the in-chain masses of the individual amino acids, permitting deduction of the sequence in conjunction with database sequence information (see Figure 3-44b, bottom panel).
Mass spectrometry is highly sensitive, able to detect as little as 1 × 10−16 mol (100 attomoles) of a peptide or 10 × 10−15 mol (10 femtomoles) of a protein of 200,000 MW. Errors in mass measurement accuracy are dependent on the specific mass analyzer used, but are typically about 0.01 percent for peptides and 0.05–0.1 percent for proteins. As described in Section 3.6, it is possible to use MS to analyze complex mixtures of proteins as well as purified proteins. MS can readily distinguish between two chemically identical peptides that differ only in that one of the peptides contains “heavy” stable (nonradioactive) isotopic forms of one or more elements (e.g., the isotopes 2H, 13C, 15N) whereas the other contains the most common, “light” isotopes (e.g., 1H, 12C, 14N) because the masses of these peptides differ. Most commonly, protein samples are digested by proteases and the peptide digestion products are subjected to analysis. An especially powerfully application of MS is to take a complex mixture of proteins from a biological specimen, digest it with trypsin or other proteases, partially separate the components using liquid chromatography, and then transfer the solution flowing out of the chromatographic column directly into an ES tandem mass spectrometer. This technique, called LC-MS/MS, which permits the nearly continuous analysis of a very complex mixture of proteins, will be described in more detail below.
The abundances of ions determined by mass spectrometry in any given sample are relative, not absolute, values. Therefore, if one wants to use MS to compare the amounts of a particular protein in two different samples (e.g., from a normal versus a mutant organism), it is necessary to have an internal standard in the samples—a molecule whose amounts do not differ between the two samples. One then determines the amounts of the protein of interest relative to that of the standard in each sample. This approach permits quantitatively accurate inter-sample comparisons of protein levels. An alternative approach involves simultaneously comparing in a single MS analysis the amounts of proteins from two different cell or tissue samples that are mixed together. This mixing approach is possible provided the proteins in one of the samples contain different stable isotopes than those in the other. The masses of the otherwise chemically identical peptides from the two samples will then differ (heavy vs. light) and can thus be distinguished by MS. Several methods can be used to chemically or enzymatically incorporate heavy or light isotopes into proteins isolated from cells for such analysis. Alternatively, cells or organisms can first be grown in the presence of amino acids containing either “heavy” or “light” isotope atoms so that these amino acids are biosynthetically incorporated into all the proteins of that sample. Cells are typically incubated with the heavy or light amino acids for five or more cell divisions to ensure that all proteins are thoroughly labeled. Proteins from the two samples are then mixed together, digested into peptides, and the peptides analyzed by mass spectrometry. Proteins and peptides derived from the “heavy” sample can be distinguished in the mass spectrometer from those from the other, “light,” sample because of their higher masses. Thus a direct comparison can be made of the relative amounts of the equivalent proteins in each sample—for example, in tumor versus normal cells or in cells treated with and without a drug. When the samples are cells grown in the laboratory, the method is called stable isotope labeling with amino acids in cell culture (SILAC).