Complete genome sequences are assembled from smaller pieces.
In Chapter 12, we discussed a method of DNA sequencing known as Sanger sequencing. Sequencing methods are now automated to the point where specialized machines can determine the sequence of billions of DNA nucleotides in a single day. However, even with recent advances, the data are obtained in the form of short sequences, typically less than a few hundred nucleotides long. If you are interested in sequencing a short DNA fragment, these technologies work well. However, let’s say you are interested in the sequence of human chromosome 1, which is a DNA molecule approximately 250 million nucleotides long. How can you sequence a DNA molecule as long as that?
In one approach, the single long DNA molecule is first broken up into small fragments, each of which is short enough to be sequenced by existing technologies. Even though the sequence data obtained from a small DNA fragment is only a minuscule fraction of the length of the DNA molecules in most genomes, each run of an automated sequencing machine yields hundreds of millions of these short sequences from random locations throughout the genome. To sequence a whole genome, researchers typically sequence such a large number of random DNA fragments that, on average, any particular small region of the genome is sequenced 10–
273
When the sequences of a sufficient number of short stretches of the genome have been obtained, the next step is sequence assembly: The short sequences are put together in the correct order to generate the long, continuous sequence of nucleotides in the DNA molecule present in each chromosome.
Assembly is accomplished by complex computer programs, but the principle is simple. The short sequences are assembled according to their overlaps, as illustrated in Fig. 13.1, which uses a sentence to represent the nucleotide sequence. This approach is called shotgun sequencing because the sequenced fragments do not originate from a particular gene or region but from sites scattered randomly across the chromosome.
Quick Check 1 DNA sequencing technology has been around since the late 1970s. Why did sequencing whole genomes present a challenge?
Quick Check 1 Answer
DNA sequencing technology is limited to DNA molecules that are much smaller than the size of a chromosome, so the challenge of genome sequencing is to piece together smaller sequenced DNA fragments.
HOW DO WE KNOW?
FIG. 13.1
How are whole genomes sequenced?
BACKGROUND DNA sequencing technologies can only determine the sequence of DNA fragments far smaller than the genome itself. How can the sequences of these small fragments be used to determine the sequence of an entire genome? In the early years of genome sequencing, many researchers thought that it would be necessary to know first where in the genome each fragment originated before sequencing it. A group at Celera Genomics reasoned that if so many fragments were sequenced that the ends of one would almost always overlap with those of others, then a computer program with sufficient power might be able to assemble the short sequences to reveal the sequence of the entire genome.
HYPOTHESIS A genome sequence can be determined by sequencing small, randomly generated DNA fragments and assembling them into a complete sequence by matching regions of overlap between the fragments.
EXPERIMENT Hundreds of millions of short sequences from the genome of the fruit fly, Drosophila melanogaster, were sequenced. Fig. 13.1a shows examples of overlapping fragments, using a sentence from Watson and Crick’s original paper on the chemical structure of DNA as an analogy.
RESULTS The computer program the group had written to assemble the fragments worked. The researchers were able to sequence the entire Drosophila genome by piecing together the fragments according to their overlaps. In the sentence analogy, the fragments (Fig. 13.1a) can be assembled into the complete sentence (Fig. 13.1b) by matching the overlaps between the fragments.
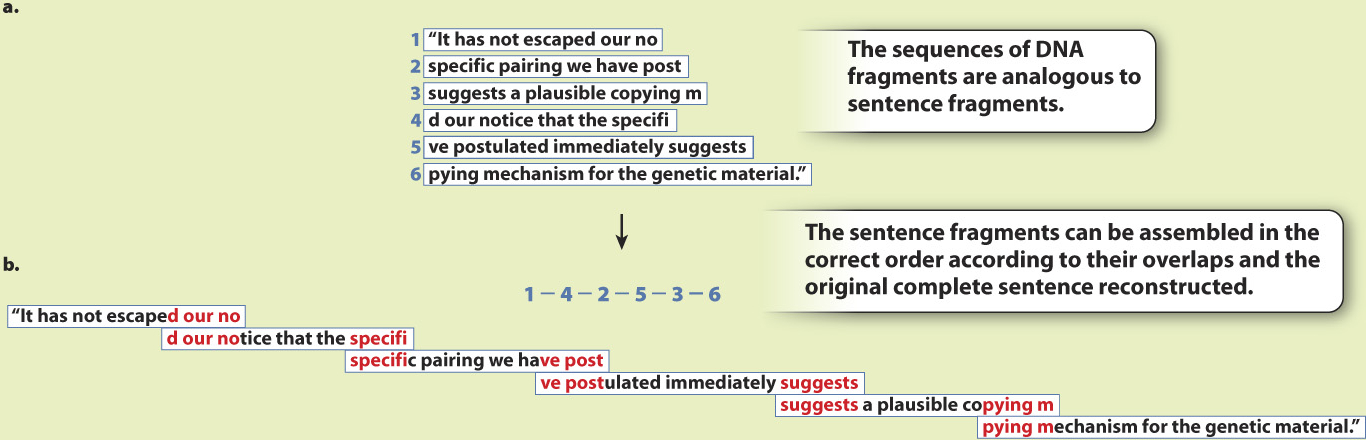
CONCLUSION The hypothesis was supported: Celera Genomics could determine the entire genomic sequence of an organism by sequencing small, random fragments and piecing them together at their overlapping ends.
FOLLOW-
SOURCE Adams, M. D., et al. 2000. “The Genome Sequence of Drosophila melanogaster.” Science 287:2185–