Sequences that are repeated complicate sequence assembly.
Sequence assembly is not quite as straightforward as Fig. 13.1 suggests. Real sequences are composed of the nucleotides A, T, G, and C, and any given short sequence could come from either strand of the double-
Some features of genomes present additional challenges to sequence assembly, and the limitations of the computer programs for handling such features require hands-
274
There are a variety of types of repeated sequence in eukaryotic genomes, and some are shown in Fig. 13.2. The repeated sequence may be several thousand nucleotides long and present in multiple identical or nearly identical copies. These long repeated sequences may be dispersed throughout the genome (Fig. 13.2a), or they may be tandem, meaning that they are next to each other (Fig. 13.2b).
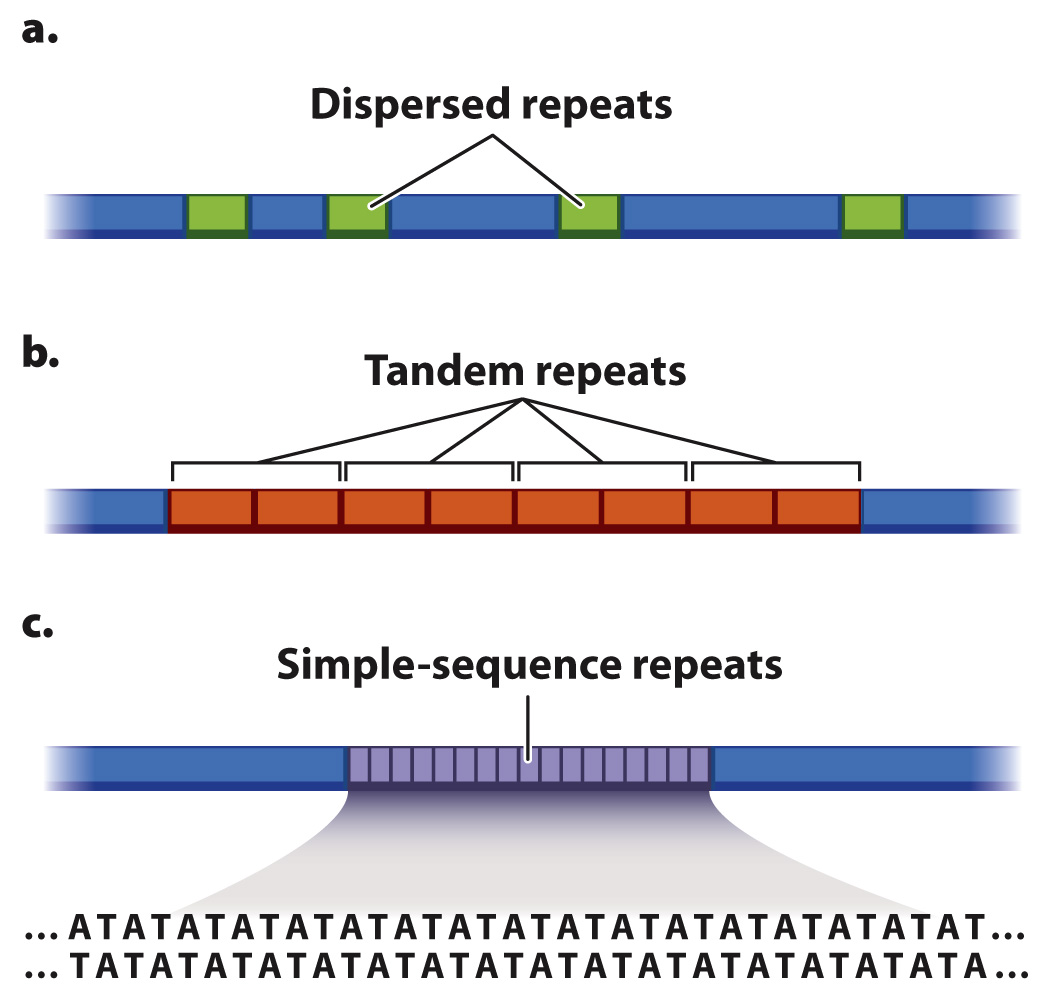
The difficulty with long repeated sequences is that they typically are much longer than the short fragments sequenced by automated sequencing. As a result, the repeat may not be detected at all. And if the repeat is detected, there is no easy way of knowing the number of copies of the repeat, that is, whether the DNA molecule includes two, three, four, or any number of copies of the repeat. Sometimes, researchers can use the ends of repeats, where the fragments overlap with an adjacent, nonrepeating sequence, as a guide to the position and number of repeats.
To illustrate the assembly problems caused by repeated sequences, let’s consider an analogy. In Shakespeare’s play Hamlet, the word “Hamlet” occurs about 500 times scattered throughout the text, like a dispersed repeat (Fig. 13.2a). If you chose short sequences of letters from the play at random and found the sequence “Hamlet” or “amlet” or “Haml,” you would have no way of knowing which of the 500 “Hamlets” these letters came from. Only if you found sequences that contained “Hamlet” overlapping with adjacent unique text could you identify their origin. A similar problem occurs in the case of tandem repeats.
In another type of repeat the repeating sequence is short, even as short as two nucleotides, such as AT, repeated over and over again in a stretch of DNA (Fig. 13.2c). Short repeating sequences of this kind are troublesome for sequencing machines because any single-
Quick Check 2 Let’s say that a stretch of repeated AT is successfully sequenced. From what you know of the difficulties of sequencing long repeated sequences, what other problems might you encounter in assembling these fragments?
Quick Check 2 Answer
Even when short repeated sequences can be sequenced, there is an assembly problem similar to that for longer repeats in which the researcher has no way of knowing where in the repeat any particular sequenced fragment should be assigned. The result is that the total number of repeats remains unresolved. It could be in the hundreds, thousands, tens of thousands, hundreds of thousands, or even millions.