18.5 A Number of Pathways Repair Changes in DNA
The integrity of DNA is under constant assault from radiation, chemical mutagens, and spontaneously arising changes. In spite of these damaging agents, the rate of mutation remains remarkably low, thanks to the efficiency with which DNA is repaired.
There are a number of complex pathways for repairing DNA, but several general statements can be made about DNA repair. First, most DNA-repair mechanisms require two nucleotide strands of DNA because most replace whole nucleotides, and a template strand is needed to specify the base sequence.
A second general feature of DNA repair is redundancy, meaning that many types of DNA damage can be corrected by more than one pathway of repair. This redundancy illustrates the extreme importance of DNA repair to the survival of the cell: if a mistake escapes one repair system, it’s likely to be repaired by another system, ensuring that almost all mistakes are corrected.
We will consider several general mechanisms of DNA repair: mismatch repair, direct repair, base-excision repair, nucleotide-excision repair, and repair of double-strand breaks.
Mismatch Repair
Replication is extremely accurate: each new copy of DNA has less than one error per billion nucleotides. However, in the process of replication, mismatched bases are incorporated into the new DNA with a frequency of about 10–4 to 10–5; so most of the errors that initially arise are corrected and never become permanent mutations. Some of these corrections are made in proofreading by the DNA polymerases.
521
Many incorrectly inserted nucleotides that escape detection by proofreading are corrected by mismatch repair. Incorrectly paired bases are detected and corrected by mismatch-repair enzymes. In addition, the mismatch-repair system corrects small unpaired loops in the DNA, such as those caused by strand slippage in replication (see Figure 18.12). Some nucleotide repeats may form secondary structures on the unpaired strand (see Figure 18.5), allowing them to escape detection by the mismatch-repair system.
After the incorporation error has been recognized, mismatch-repair enzymes cut out a section of the newly synthesized strand and fill the gap with new nucleotides by using the original DNA strand as a template. For this strategy to work, mismatch repair must have some way of distinguishing between the old and the new strands of the DNA so that the incorporation error, but not part of the original strand, is removed.
The proteins that carry out mismatch repair in E. coli differentiate between old and new strands by the presence of methyl groups on special sequences of the old strand. After replication, adenine nucleotides in the sequence GATC are methylated. The process of methylation is delayed and so, immediately after replication, the old strand is methylated and the new strand is not (Figure 18.36a). The mismatch-repair complex brings an unmethylated GATC sequence in close proximity to the mismatched bases. It nicks the unmethylated strand at the GATC site (Figure 18.36b), and degrades the strand between the nick and the mismatched bases (Figure 18.36c). DNA polymerase and DNA ligase fill in the gap on the unmethylated strand with correctly paired nucleotides (Figure 18.36d).
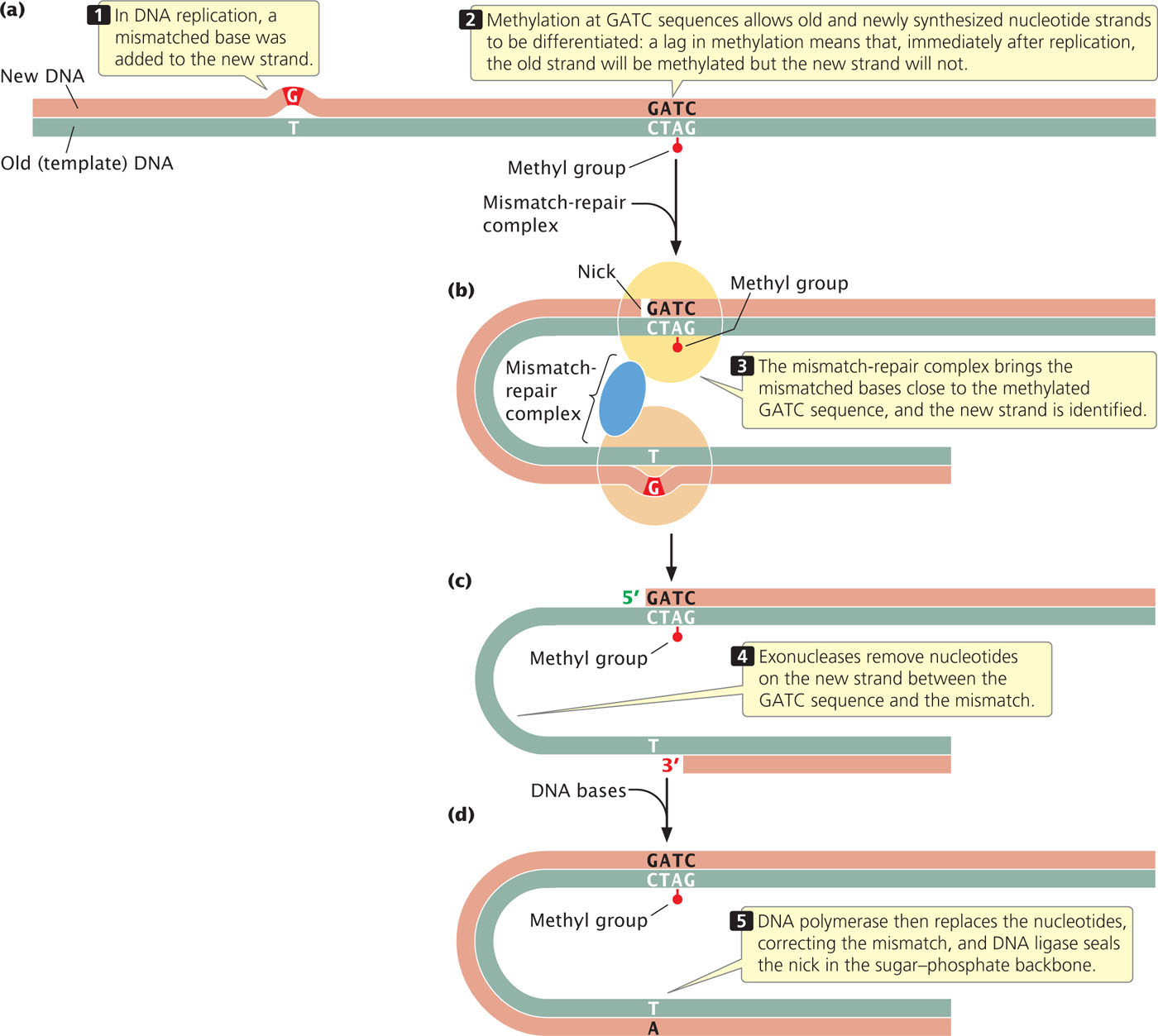
Mismatch repair in eukaryotic cells is similar to that in E. coli, but how the old and new strands are recognized in eukaryotic cells is not known. In some eukaryotes, such as yeast and fruit flies, there is no detectable methylation of DNA, and yet mismatch repair still takes place. Humans who possess mutations in mismatch-repair genes often exhibit elevated somatic mutations and are frequently susceptible to colon cancer.
CONCEPTS
Mismatched bases and other DNA lesions are corrected by mismatch repair. Enzymes cut out a section of the newly synthesized strand of DNA and replace it with new nucleotides.
 CONCEPT CHECK 9
CONCEPT CHECK 9
Mismatch repair in E. coli distinguishes between old and new strands of DNA on the basis of
- differences in base composition of the two strands.
- modification of histone proteins.
- base analogs on the new strand.
- methyl groups on the old strand.
522
Direct Repair
Direct repair does not replace altered nucleotides but, instead, changes them back into their original (correct) structures. One of the best understood direct-repair mechanisms is the photoreactivation of UV-induced pyrimidine dimers (see Figure 18.21). E. coli and some eukaryotic cells possess an enzyme called photolyase, which uses energy captured from light to break the covalent bonds that link the pyrimidines in a dimer.
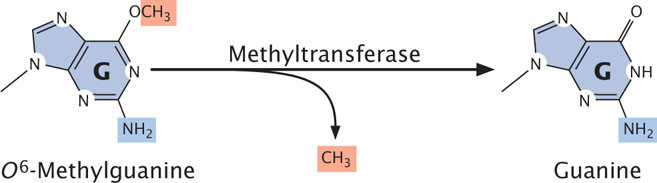
Direct repair also corrects O6-methylguanine, an alkylation product of guanine that pairs with adenine, producing G · C → T · A transversions. An enzyme called O6-methylguanine-DNA methyltransferase removes the methyl group from O6-methylguanine, restoring the base to guanine (Figure 18.37).
Base-Excision Repair
In base-excision repair, a modified base is first excised and then the entire nucleotide is replaced. The excision of modified bases is catalyzed by a set of enzymes called DNA glycosylases, each of which recognizes and removes a specific type of modified base by cleaving the bond that links that base to the 1′-carbon atom of deoxyribose sugar (Figure 18.38a). Uracil glycosylase, for example, recognizes and removes uracil produced by the deamination of cytosine. Other glycosylases recognize hypoxanthine, 3-methyladenine, 7-methylguanine, and other modified bases.
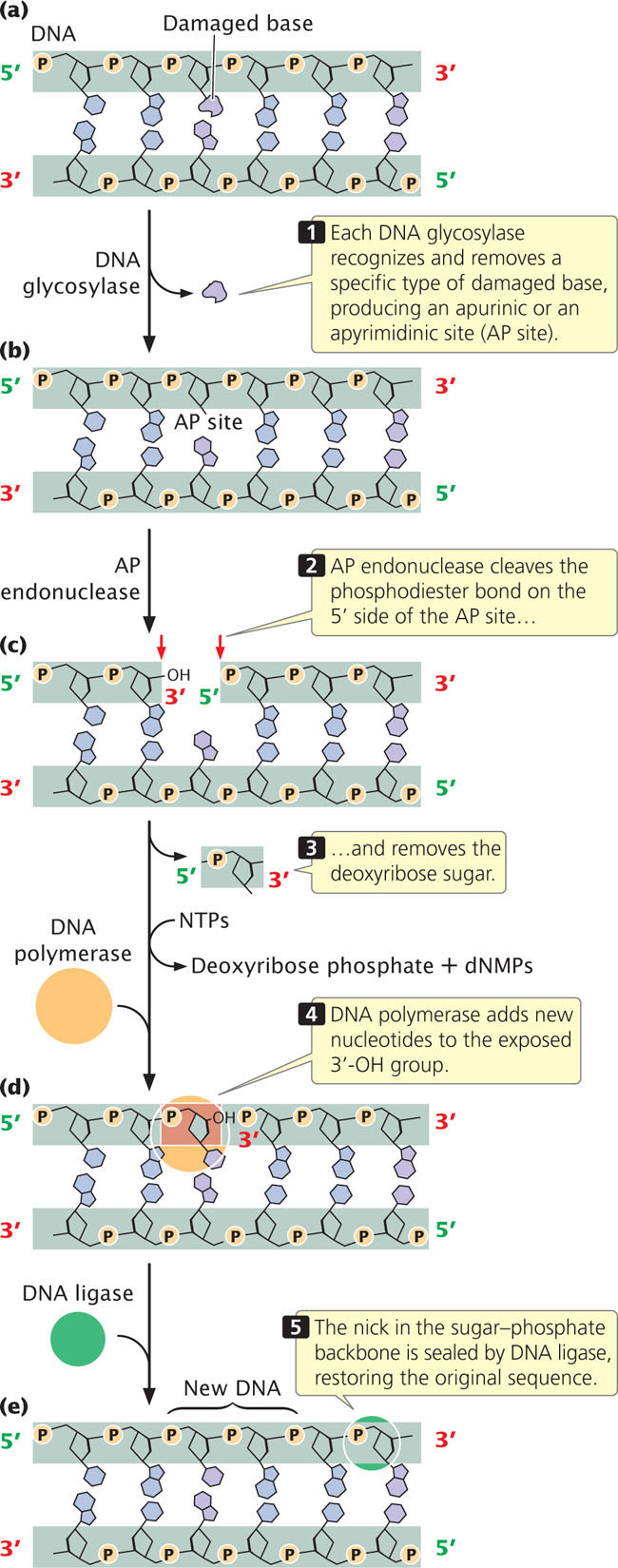
After the base has been removed, an enzyme called AP (apurinic or apyrimidinic) endonuclease cuts the phosphodiester bond, and other enzymes remove the deoxyribose sugar (Figure 18.38b). DNA polymerase then adds one or more new nucleotides to the exposed 3′-OH group (Figure 18.38c), replacing a section of nucleotides on the damaged strand. The nick in the phosphodiester backbone is sealed by DNA ligase (Figure 18.38d), and the original intact sequence is restored (Figure 18.38e).
Bacteria use DNA polymerase I to replace excised nucleotides, but eukaryotes use DNA polymerase β, which has no proofreading ability and tends to make mistakes. On average, DNA polymerase β makes one mistake per 4000 nucleotides inserted. About 20,000 to 40,000 base modifications per day are repaired by base excision, and so DNA polymerase β may introduce as many as 10 mutations per day into the human genome. How are these errors corrected? Recent research results show that some AP endonucleases have the ability to proofread. When DNA polymerase β inserts a nucleotide with the wrong base into the DNA, DNA ligase cannot seal the nick in the sugar–phosphate backbone, because the 3′-OH and 5′-P groups of adjacent nucleotides are not in the correct orientation for ligase to connect them. In this case, AP endonuclease 1 detects the mispairing and uses its 3′→5′ exonuclease activity to excise the incorrectly paired base. DNA polymerase β then uses its polymerase activity to fill in the missing nucleotide. In this way, the fidelity of base-excision repair is maintained.
523
CONCEPTS
Direct-repair mechanisms change altered nucleotides back into their correct structures. In base-excision repair, glycosylase enzymes recognize and remove specific types of modified bases. The entire nucleotide is then removed and a section of the polynucleotide strand is replaced.
CONCEPT CHECK 10
How do direct-repair mechanisms differ from mismatch repair and base-excision repair?
Nucleotide-Excision Repair
Another repair pathway is nucleotide-excision repair, which removes bulky DNA lesions (such as pyrimidine dimers) that distort the double helix. Nucleotide-excision repair can repair many different types of DNA damage and is found in cells of all organisms from bacteria to humans.
The process of nucleotide excision is complex; in humans, a large number of genes take part. First, a complex of enzymes scans DNA, looking for distortions of its three-dimensional configuration (Figure 18.39a and b). When a distortion is detected, additional enzymes separate the two nucleotide strands at the damaged region, and single-strand-binding proteins stabilize the separated strands (Figure 18.39c). Next, the sugar–phosphate backbone of the damaged strand is cleaved on both sides of the damage (Figure 18.39d). Part of the damaged strand is peeled away by helicase enzymes (Figure 18.39e), and the gap is filled in by DNA polymerase and sealed by DNA ligase (Figure 18.39f).
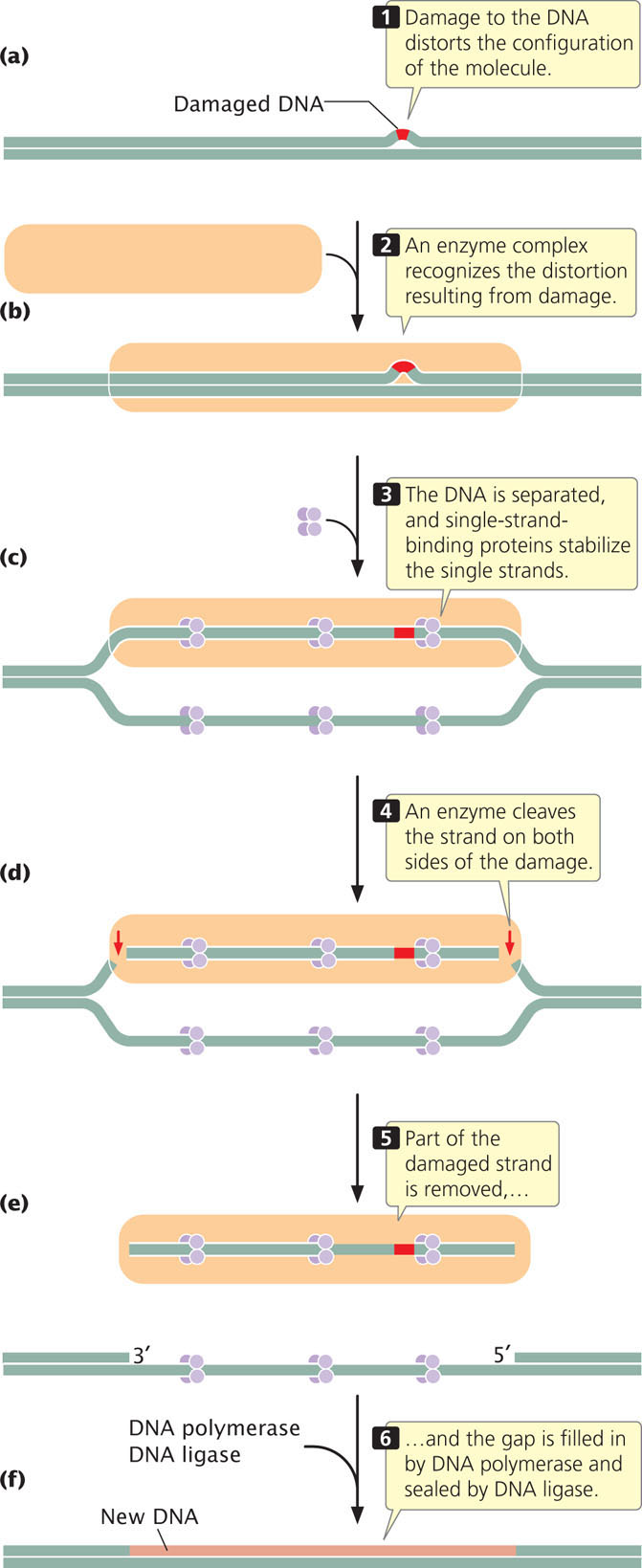
CONCEPTS
Nucleotide-excision repair removes and replaces many types of damaged DNA that distort the DNA structure. The two strands of DNA are separated, a section of the DNA containing the distortion is removed, DNA polymerase fills in the gap, and DNA ligase seals the filled-in gap.
524
CONNECTING CONCEPTS: The Basic Pathway of DNA Repair
We have now examined several different mechanisms of DNA repair. What do these methods have in common? How are they different? Most methods of DNA repair depend on the presence of two strands, because nucleotides in the damaged area are removed and replaced. Nucleotides are replaced in mismatch repair, base-excision repair, and nucleotide-excision repair but are not replaced by direct-repair mechanisms.
Repair mechanisms that include nucleotide removal utilize a common four-step pathway:
- Detection: The damaged section of the DNA is recognized.
- Excision: DNA-repair endonucleases nick the phosphodiester backbone on one or both sides of the DNA damage and one or more nucleotides are removed.
- Polymerization: DNA polymerase adds nucleotides to the newly exposed 3′-OH group by using the other strand as a template and replacing damaged (and frequently some undamaged) nucleotides.
- Ligation: DNA ligase seals the nicks in the sugar–phosphate backbone.
The primary differences in the mechanisms of mismatch, base-excision, and nucleotide-excision repair are in the details of detection and excision. In base-excision and mismatch repair, a single nick is made in the sugar–phosphate backbone on one side of the damage; in nucleotide-excision repair, nicks are made on both sides of the DNA lesion. In base-excision repair, DNA polymerase displaces the old nucleotides as it adds new nucleotides to the 3′ end of the nick; in mismatch repair, the old nucleotides are degraded; and, in nucleotide-excision repair, nucleotides are displaced by helicase enzymes. All three mechanisms use DNA polymerase and ligase to fill in the gap produced by the excision and removal of damaged nucleotides.
Repair of Double-Strand Breaks
A common type of DNA damage is a double-strand break, in which both strands of the DNA helix are broken. Double-strand breaks are caused by ionizing radiation, oxidative free radicals, and other DNA-damaging agents. These types of breaks are particularly detrimental to the cell because they stall DNA replication and may lead to chromosome rearrangements, such as deletions, duplications, inversions, and translocations. There are two major pathways for repairing double-strand breaks: homologous recombination and nonhomologous end joining.
Homologous Recombination
Homologous recombination repairs a broken DNA molecule by using the identical or nearly identical genetic information contained in another DNA molecule, usually a sister chromatid—the same mechanism employed in the process of homologous recombination that is responsible for crossing over (see Chapter 12). Homologous recombination begins with the removal of some nucleotides at the broken ends, followed by strand invasion, displacement, and replication (see Figure 12.20). Many of the same enzymes that carry out crossing over are utilized in the repair of double-strand breaks by homologous recombination: two such enzymes are BRCA1 and BRCA2. The genes that code for these proteins are frequently mutated in breast cancer cells.
Nonhomologous End Joining
Nonhomologous end joining repairs double-strand breaks without using a homologous template. This pathway is often used when the cell is in G1 and a sister chromatid is not available for repair through homologous recombination. Nonhomologous end joining uses proteins that recognize the broken ends of DNA, bind to the ends, and then joins them together. Nonhomologous end joining is more error prone than homologous recombination and often leads to deletions, insertions, and translocations. Different types of DNA repair are summarized in Table 18.5.
| Repair System | Type of Damage Repaired |
|---|---|
| Mismatch | Replication errors, including mispaired bases and strand slippage |
| Direct | Pyrimidine dimers; other specific types of alterations |
| Base excision | Abnormal bases, modified bases, and pyrimidine dimers |
| Nucleotide excision | DNA damage that distorts the double helix, including abnormal bases, modified bases, and pyrimidine dimers |
| Homologous recombination | Double-strand breaks |
| Nonhomologous end joining | Double-strand breaks |
Translesion DNA Polymerases
As discussed in Chapter 12, the high-fidelity DNA polymerases that normally carry out replication operate at high speed and, like a high-speed train, require a smooth track—an undistorted template. Some mutations, such as pyrimidine dimers, produce distortions in the three-dimensional structure of the DNA helix, blocking replication by the high-speed polymerases. When distortions of the template are encountered, specialized translesion DNA polymerases take over replication and bypass the lesions.
525
The translesion polymerases are able to bypass bulky lesions but, in the process, often make errors. Thus, the translesion polymerases allow replication to proceed at the cost of introducing mutations into the sequence. Some of these mutations are corrected by DNA-repair systems, but others escape detection.
An example of a translesion DNA polymerase is polymerase η (eta), which bypasses pyrimidine dimers in eukaryotes. Polymerase η inserts AA opposite a pyrimidine dimer. This strategy seems to be reasonable because about two-thirds of pyrimidine dimers are thymine dimers. However, the insertion of AA opposite a CT dimer results in a C · G → T · A transversion. Polymerase η therefore tends to introduce mutations into the DNA sequence.
CONCEPTS
Two major pathways exist for the repair of double-strand breaks in DNA: homologous recombination and nonhomologous end joining. Special translesion DNA polymerases allow replication to proceed past bulky distortions in the DNA but often introduce errors as they bypass the distorted region.
Genetic Diseases and Faulty DNA Repair
Several human diseases are connected to defects in DNA repair. These diseases are often associated with high incidences of specific cancers, because defects in DNA repair lead to increased rates of mutation. This concept is discussed further in Chapter 23.
Among the best studied of the human DNA-repair diseases is xeroderma pigmentosum (Figure 18.40), a rare autosomal recessive condition that includes abnormal skin pigmentation and acute sensitivity to sunlight. Persons who have this disease also have a strong predisposition to skin cancer, with an incidence ranging from 1000 to 2000 times that found in unaffected people.

Sunlight includes a strong UV component, so exposure to sunlight produces pyrimidine dimers in the DNA of skin cells. Although human cells lack photolyase (the enzyme that repairs pyrimidine dimers in bacteria), most pyrimidine dimers in humans can be corrected by nucleotide-excision repair (see Figure 18.39). However, the cells of most people with xeroderma pigmentosum are defective in nucleotide-excision repair, and many of their pyrimidine dimers go uncorrected and may lead to cancer.
Xeroderma pigmentosum can result from defects in several different genes. Some persons with xeroderma pigmentosum have mutations in a gene encoding the protein that recognizes and binds to damaged DNA; others have mutations in a gene encoding helicase. Still others have defects in the genes that play a role in cutting the damaged strand on the 5′ or 3′ sides of the pyrimidine dimer. Some persons have a slightly different form of the disease (xeroderma pigmentosum variant) owing to mutations in the gene encoding polymerase η, the translesion DNA polymerase that bypasses pyrimidine dimers.
Another genetic disease caused by faulty DNA repair is an inherited form of colon cancer called hereditary nonpolyposis colon cancer (HNPCC). This is one of the most common hereditary cancers, accounting for about 15% of colon cancers. Research findings indicate that HNPCC arises from mutations in the proteins that carry out mismatch repair (see Figure 18.36). Some genetic diseases associated with defective DNA repair are summarized in Table 18.6. ![]() TRY PROBLEM 44
TRY PROBLEM 44
| Disease | Symptoms | Genetic Defect |
|---|---|---|
| Xeroderma pigmentosum | Frecklelike spots on skin, sensitivity to sunlight, predisposition to skin cancer | Defects in nucleotide-excision repair |
| Cockayne syndrome | Dwarfism, sensitivity to sunlight, premature aging, deafness, intellectual disability | Defects in nucleotide-excision repair |
| Trichothiodystrophy | Brittle hair, skin abnormalities, short stature, immature sexual development, characteristic facial features | Defects in nucleotide-excision repair |
| Hereditary nonpolyposis colon cancer | Predisposition to colon cancer | Defects in mismatch repair |
| Fanconi anemia | Increased skin pigmentation, abnormalities of skeleton, heart, and kidneys, predisposition to leukemia | Possibly defects in the repair of interstrand cross-links |
| Li-Fraumeni syndrome | Predisposition to cancer in many different tissues | Defects in DNA damage response |
| Werner syndrome | Premature aging, predisposition to cancer | Defect in homologous recombination |
CONCEPTS
Defects in DNA repair are the underlying cause of several genetic diseases. Many of these diseases are characterized by a predisposition to cancer.
CONCEPT CHECK 11
Why are defects in DNA repair often associated with increases in cancer?
526