19.2 Molecular Techniques Are Used to Isolate, Recombine, and Amplify Genes
A first step in the molecular analysis of a DNA segment or gene is to isolate it from the remainder of the DNA and to make many copies of it so that further analyses can be carried out. The isolation and amplification of DNA frequently requires that it be recombined with other DNA molecules. In the sections that follow, we will examine some of the molecular techniques that are used to isolate, recombine, and amplify DNA segments.
Cutting and Joining DNA Fragments
A key event in the development of molecular genetic methods was the discovery in the late 1960s of restriction enzymes (also called restriction endonucleases) that recognize and make double-stranded cuts in DNA at specific nucleotide sequences. These enzymes are produced naturally by bacteria and are used in defense against viruses. A bacterium protects its own DNA from a restriction enzyme by modifying the recognition sequence, usually by adding methyl groups to its DNA.
Several different types of restriction enzymes have been isolated from bacteria. Type II restriction enzymes recognize specific sequences and cut the DNA at defined sites within or near the recognition sequence. Virtually all molecular genetics work is done with type II restriction enzymes; discussions of restriction enzymes throughout this book refer to type II enzymes.
More than 800 different restriction enzymes that recognize and cut DNA at more than 100 different sequences have been isolated from bacteria. Many of these enzymes are commercially available; examples of some commonly used restriction enzymes are given in Table 19.1. The name of each restriction enzyme begins with an abbreviation that signifies its bacterial origin.
The sequences recognized by restriction enzymes are usually from 4 to 8 bp long; most enzymes recognize a sequence of 4 or 6 bp. Most recognition sequences are palindromic—sequences that read the same (5′ to 3′) on the two complementary DNA strands.
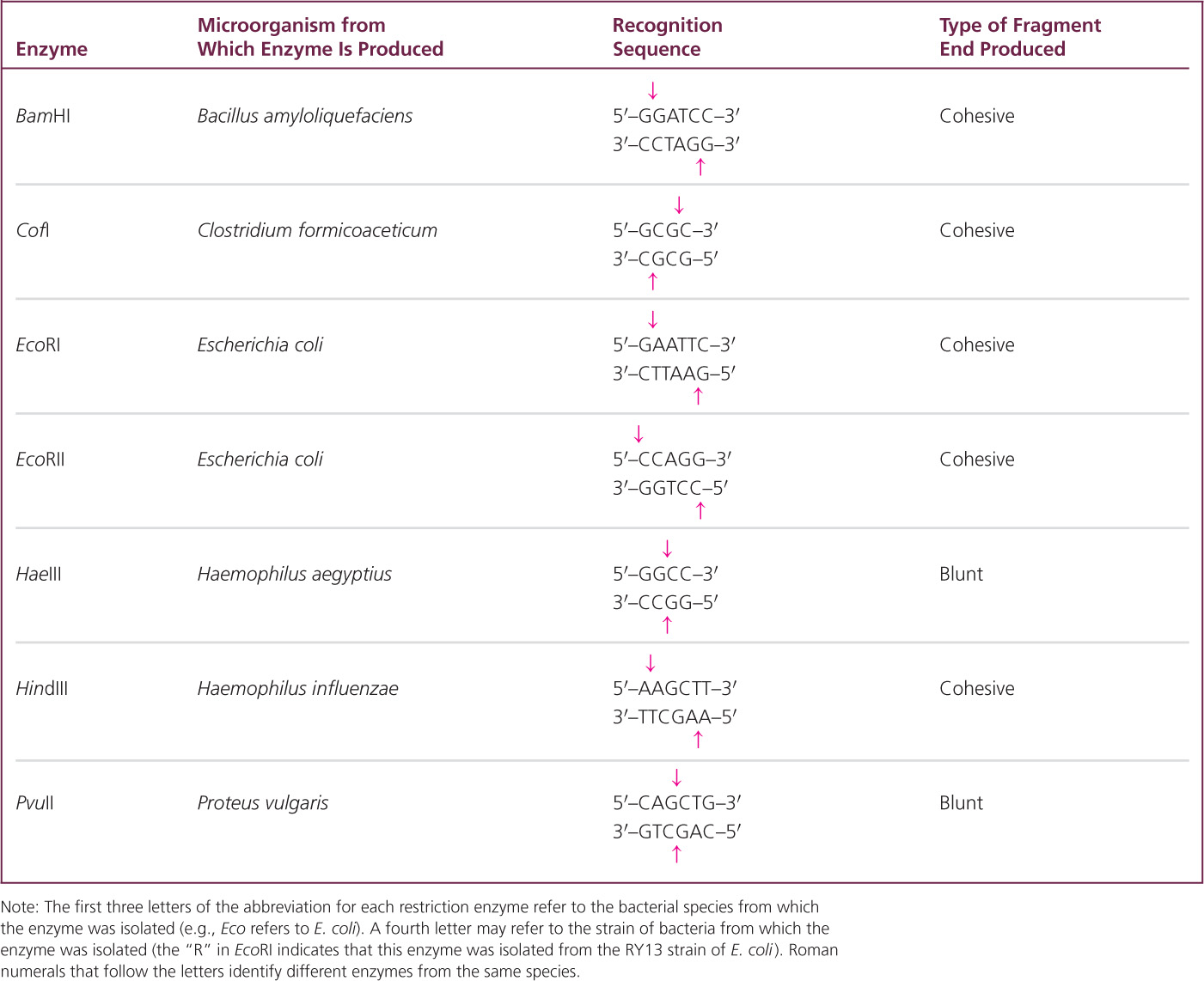
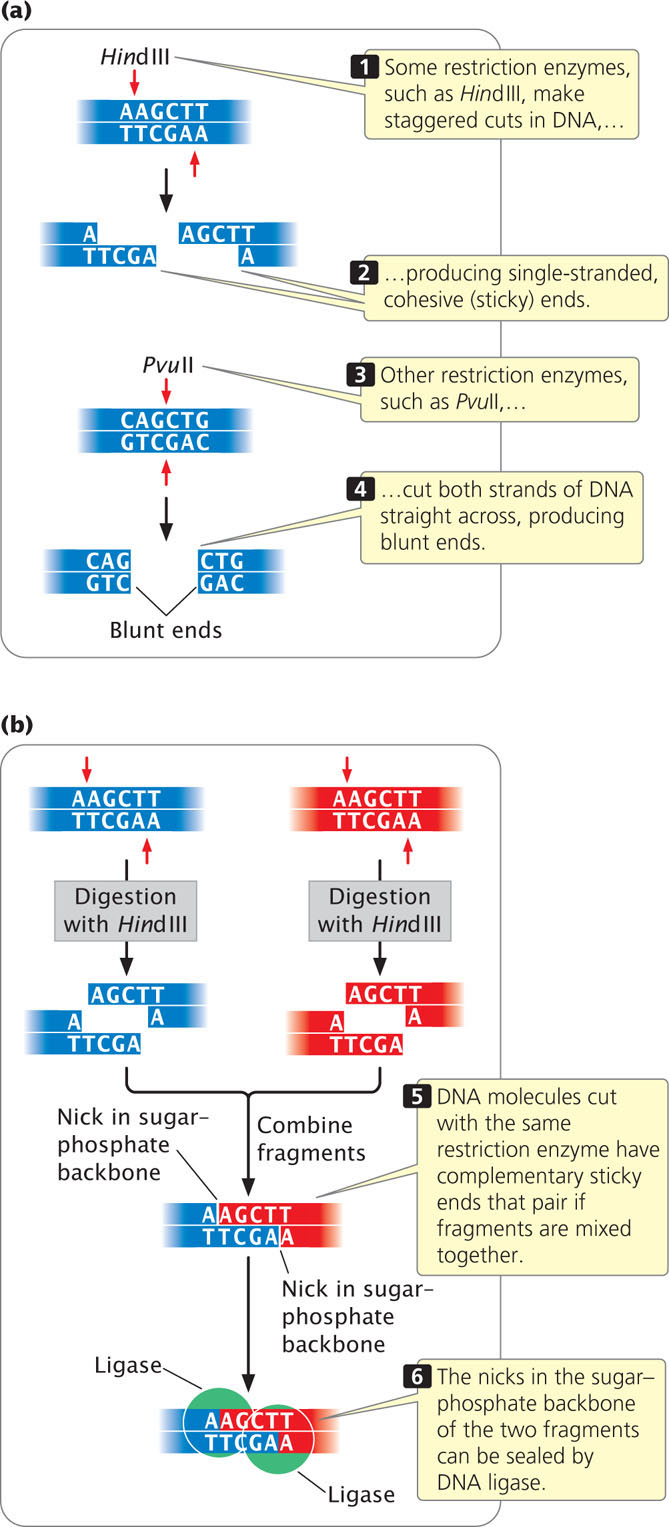
Some of the enzymes make staggered cuts in the DNA. For example, HindIII recognizes the following sequence:

HindIII cuts the sugar–phosphate backbone of each strand at the point indicated by the arrow, generating fragments with short, single-stranded overhanging ends:
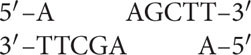
Such ends are called cohesive ends or sticky ends because they are complementary to each other and can spontaneously pair to connect the fragments. Thus, DNA fragments can be “glued” together: any two fragments cleaved by the same enzyme will have complementary ends and will pair (Figure 19.2). When their cohesive ends have paired, two DNA fragments can be joined together permanently by DNA ligase, which seals nicks between the sugar–phosphate groups of the fragments.
538
Not all restriction enzymes produce staggered cuts and sticky ends. PvuII cuts in the middle of its recognition site and the cuts on the two strands are directly opposite one another, producing blunt-ended fragments:
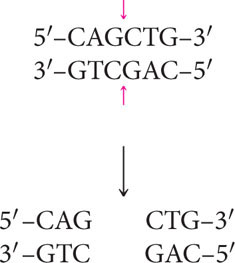
Fragments that have blunt ends are usually joined together in other ways.
The sequences recognized by a restriction enzyme are located randomly within the genome. Accordingly, there is a relation between the length of the recognition sequence and the number of times that it is present in a genome: there will be fewer longer recognition sequences than shorter recognition sequences because the probability of the occurrence of a particular sequence consisting of, say, six specific bases is less than the probability of the occurrence of a particular sequence of four specific bases. Consequently, restriction enzymes that recognize longer sequences will cut a given piece of DNA into fewer and longer fragments than will restriction enzymes that recognize shorter sequences.
Restriction enzymes are used whenever DNA fragments must be cut or joined. In a typical restriction reaction, a concentrated solution of purified DNA is placed in a small tube with a buffer solution and a small amount of restriction enzyme. The reaction mixture is then heated at the optimal temperature for the enzyme, usually 37°C. Within a few hours, the enzyme cuts the appropriate restriction sites in all the DNA molecules, producing a set of DNA fragments.
539
In recent years, geneticists have designed more complex enzymes, termed engineered nucleases, which are capable of making double-stranded cuts to the DNA at any predetermined DNA sequence. Engineered nucleases consist of part of a restriction enzyme that cleaves the DNA, coupled with another protein that recognizes and binds to a specific DNA sequence; the particular sequence to which the protein binds is determined by the protein’s amino acid sequence. By altering the amino acid sequence of the binding protein, the engineered nuclease can be custom designed to bind to and cut any particular DNA sequence. Engineered nucleases include zinc finger nucleases (ZFNs), which use a DNA-binding protein called a zinc finger, and transcription activator-like effector nucleases (TALENs), which use a protein that normally binds to sequences in promoters. ![]() TRY PROBLEM 29
TRY PROBLEM 29
CONCEPTS
Type II restriction enzymes cut DNA at specific base sequences that are palindromic. Some restriction enzymes make staggered cuts, producing DNA fragments with cohesive ends; others cut both strands straight across, producing blunt-ended fragments. There are fewer long recognition sequences in DNA than short recognition sequences.
 CONCEPT CHECK 1
CONCEPT CHECK 1
Where do restriction enzymes come from?
Viewing DNA Fragments
After the completion of a restriction reaction, a number of questions arise. Did the restriction enzyme cut the DNA? Into how many fragments was the DNA cut? What are the sizes of the resulting fragments? Gel electrophoresis provides us with a means of answering these questions.
Electrophoresis is a standard biochemical technique for separating molecules on the basis of their size and electrical charge. There are a number of different types of electrophoresis; to separate DNA molecules, gel electrophoresis is used. A porous gel is often made from agarose (a polysaccharide isolated from seaweed), which is melted in a buffer solution and poured into a plastic mold. As it cools, the agarose solidifies, making a gel that looks something like stiff gelatin.
Small wells are made at one end of the gel to hold solutions of DNA fragments (Figure 19.3a), and an electrical current is passed through the gel. Because the phosphate group of each DNA nucleotide carries a negative charge, the DNA fragments migrate toward the positive end of the gel. In this migration, the porous gel acts as a sieve, separating the DNA fragments by size. Small DNA fragments migrate more rapidly than do large ones and, with the passage of time, the fragments separate on the basis of their size. Typically, DNA fragments of known length (size standards) are placed in another well. By comparing the migration distance of the unknown fragments with the distance traveled by the size standards, a researcher can determine the approximate size of the unknown fragments (Figure 19.3b).
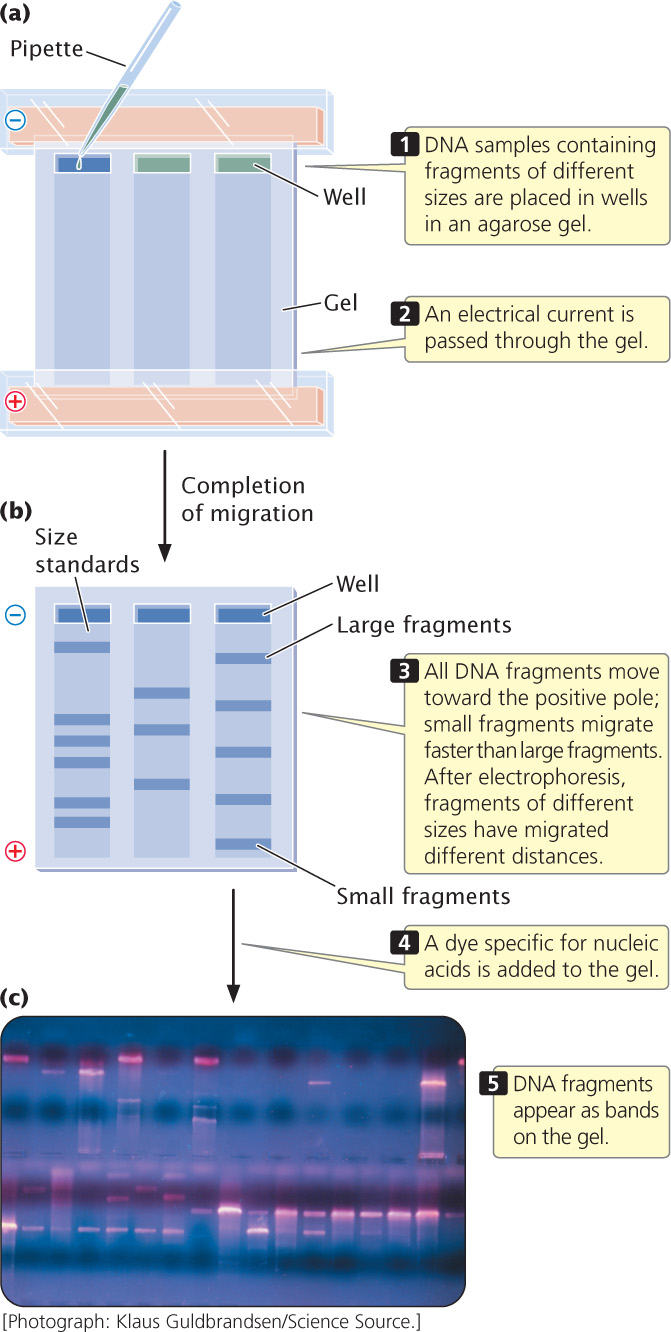
The DNA fragments are still too small to see, so the problem of visualizing the DNA needs to be addressed. Visualization can be accomplished in several ways. The simplest procedure is to stain the gel with a dye specific for nucleic acids, such as ethidium bromide, which wedges itself tightly (intercalates) between the bases of DNA and fluoresces when exposed to UV light, producing brilliant bands on the gel (Figure 19.3c). Alternatively, DNA fragments can be visualized by adding a label to the DNA before it is placed in the gel. For example, chemical labels can be detected by adding antibodies or other substances that carry a dye and will attach to the relevant DNA, which can then be visualized directly. ![]() TRY PROBLEM 30
TRY PROBLEM 30
540
CONCEPTS
DNA fragments can be separated, and their sizes can be determined with the use of gel electrophoresis. The fragments can be viewed by using a dye that is specific for nucleic acids or by labeling the fragments with a chemical tag.
CONCEPT CHECK 2
DNA fragments that are 500 bp, 1000 bp, and 2000 bp in length are separated by gel electrophoresis. Which fragment will migrate farthest in the gel?
- 2000-bp fragment
- 1000-bp fragment
- 500-bp fragment
- All will migrate equal distances.
Locating DNA Fragments with Southern Blotting and Probes
If a small piece of DNA, such as a plasmid, is cut by a restriction enzyme, the few fragments produced can be seen as distinct bands on an electrophoretic gel. In contrast, if genomic DNA from a cell is cut by a restriction enzyme, a large number of fragments of different sizes are produced. A restriction enzyme that recognizes a four-base sequence would theoretically cut about once every 256 bp. The human genome, with 3.2 billion base pairs, would generate more than 12 million fragments when cut by this restriction enzyme. Separated by electrophoresis and stained, this large set of fragments would appear as a continuous smear on the gel because of the presence of so many fragments of differing size. Usually, researchers are interested in only a few of these fragments, perhaps those carrying a specific gene. How can they locate the desired fragments in such a large pool of DNA?
One approach is to use a probe, which is a DNA or RNA molecule with a base sequence complementary to a sequence in the gene of interest. The bases on a probe will pair only with the bases on a complementary sequence and, if suitably labeled, the probe can be used to locate a specific gene or other DNA sequence. To use a probe, a researcher first cuts the DNA into fragments by using one or more restriction enzymes and then separates the fragments with gel electrophoresis (Figure 19.4). Next, the separated fragments must be denatured and transferred to a permanent solid medium (such as nitrocellulose or nylon membrane). Southern blotting (named after Edwin M. Southern) is one technique used to transfer the denatured, single-stranded fragments from a gel to a permanent solid medium.
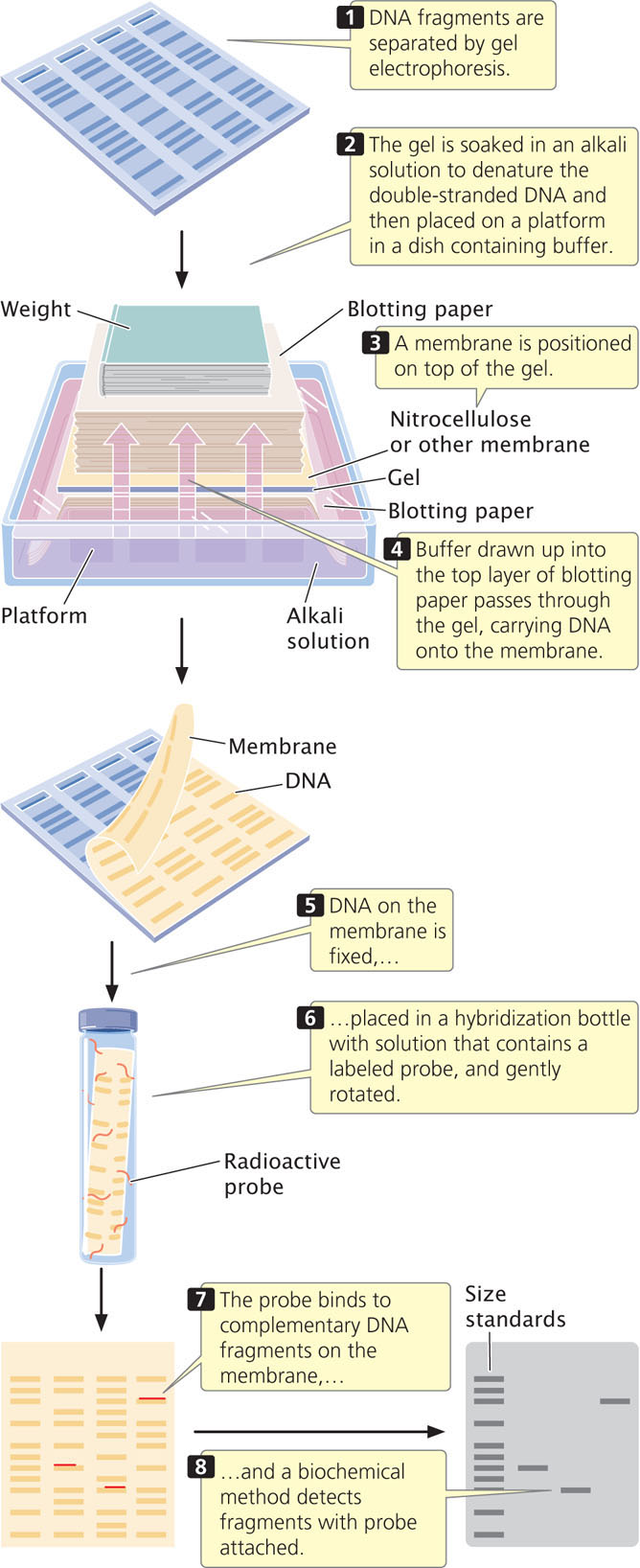
After the single-stranded DNA fragments have been transferred, the membrane is placed in a hybridization solution of a labeled probe (see Figure 19.4). The probe will bind to any DNA fragments on the membrane that bear complementary sequences. Often, a probe binds to only a part of the DNA fragment, and so the DNA fragment may contain sequences not found in the probe. The membrane is then washed to remove any unbound probe; a biochemical method reveals the presence of the bound probe.
541
RNA can be transferred from a gel to a solid support by a related procedure called Northern blotting (not named after anyone but capitalized to match Southern). The hybridization of a probe can reveal the size of a particular mRNA molecule, its relative abundance, or the tissues in which the mRNA is transcribed. Western blotting is the transfer of protein from a gel to a membrane. Here, the probe is usually an antibody, used to determine the size of a particular protein and the pattern of the protein’s expression.
CONCEPTS
Labeled probes, which are sequences of RNA or DNA that are complementary to the sequence of interest, can be used to locate individual genes or DNA sequences. Southern blotting can be used to transfer DNA fragments from a gel to a membrane such as nitrocellulose.
CONCEPT CHECK 3
How do Northern and Western blotting differ from Southern blotting?
Cloning Genes
Many recombinant DNA methods require numerous copies of a specific DNA fragment. One way to amplify a specific piece of DNA is to place the fragment in a bacterial cell and allow the cell to replicate the DNA. This procedure is termed gene cloning because identical copies (clones) of the original piece of DNA are produced.
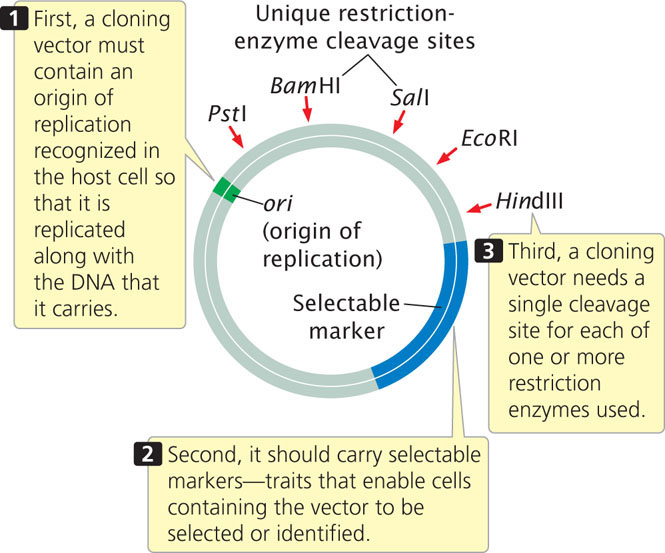
A cloning vector is a stable, replicating DNA molecule to which a foreign DNA fragment can be attached for introduction into a cell. An effective cloning vector has three important characteristics (Figure 19.5): (1) an origin of replication, which ensures that the vector is replicated within the cell; (2) selectable markers, which enable any cells containing the vector to be selected or identified; and (3) one or more unique restriction sites into which a DNA fragment can be inserted. The restriction sites used for cloning must be unique; if a vector is cut at multiple recognition sites, several pieces of DNA are generated and getting these back together in the correct order is possible but extremely difficult.
542
Plasmid Vectors
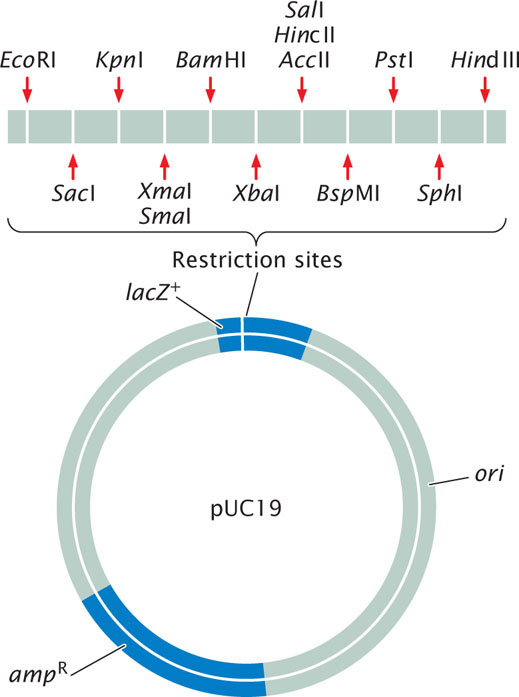
Plasmids, circular DNA molecules that exist naturally in bacteria (see Chapter 9), are commonly used vectors for cloning DNA fragments in bacteria. They contain origins of replication and are therefore able to replicate independently of the bacterial chromosome. The plasmids typically used in cloning have been constructed from the larger, naturally occurring bacterial plasmids and have multiple restriction sites, an origin of replication site, and selectable markers (Figure 19.6).
The easiest method for inserting a gene into a plasmid vector is to cut the foreign DNA (containing the gene) and the plasmid with the same restriction enzyme (Figure 19.7). If the restriction enzyme makes staggered cuts in the DNA, complementary sticky ends are produced on the foreign DNA and the plasmid. The DNA and plasmid are then mixed together; some of the foreign DNA fragments will pair with the cut ends of the plasmid. DNA ligase is used to seal the nicks in the sugar–phosphate backbone, creating a recombinant plasmid that contains the foreign DNA fragment. You can learn more about plasmid cloning by viewing  Animation 19.1.
Animation 19.1.
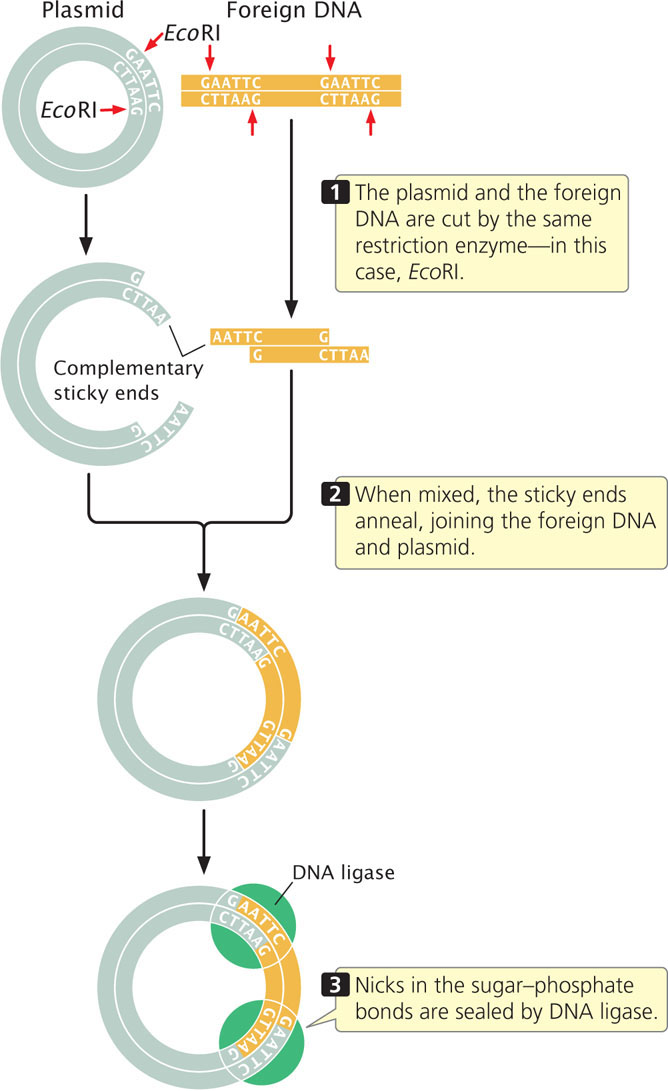
Sometimes restriction sites are not available at a site where the DNA needs to be cut. In that case, a restriction site can be created with the use of linkers, which are small, synthetic DNA fragments that contain one or more restriction sites. Linkers can be attached to the ends of any piece of DNA and are then cut by a restriction enzyme, generating sticky ends that are complementary to sticky ends on the plasmid.
Transformation
When a gene has been placed inside a plasmid, the plasmid must be introduced into bacterial cells. This task is usually accomplished by transformation, in which bacterial cells take up DNA from the external environment (see Chapter 9). Some types of cells undergo transformation naturally; others must be treated chemically or physically before they will undergo transformation. Inside the cell, the plasmids replicate and multiply, and the cells themselves multiply.
Screening Cells for Recombinant Plasmids
Cells bearing recombinant plasmids can be detected with the use of the selectable markers on the plasmid. Common selectable markers are genes that confer resistance to an antibiotic: any cell that contains such a plasmid will be able to live in the presence of the antibiotic, which normally kills bacterial cells.
543
A common way to screen cells for the presence of a recombinant plasmid is to use a plasmid that contains a fragment of the lacZ gene—a small part of the front end of the gene (Figure 19.8). This partial lacZ gene contains a series of unique restriction sites into which a piece of DNA can be inserted and cloned. The bacteria that will do the work of cloning have special features that make the presence of the recombinant plasmid evident. The bacteria that are to be transformed by the plasmid are lacZ−, missing the front end of the lacZ gene but containing its back end. Without the plasmid the bacteria are unable to synthesize β-galactosidase. If no foreign DNA has been inserted into the partial lacZ gene of the plasmid, the front end of the gene (provided by the plasmid) and back end of the gene (provided by the bacteria) work together within the bacterial cell to produce β-galactosidase. If foreign DNA is successfully inserted into the restriction site, it disrupts the front end of the lacZ gene, and β-galactosidase is not produced. The plasmid also usually contains a selectable marker, which may be a gene that confers resistance to an antibiotic such as ampicillin.
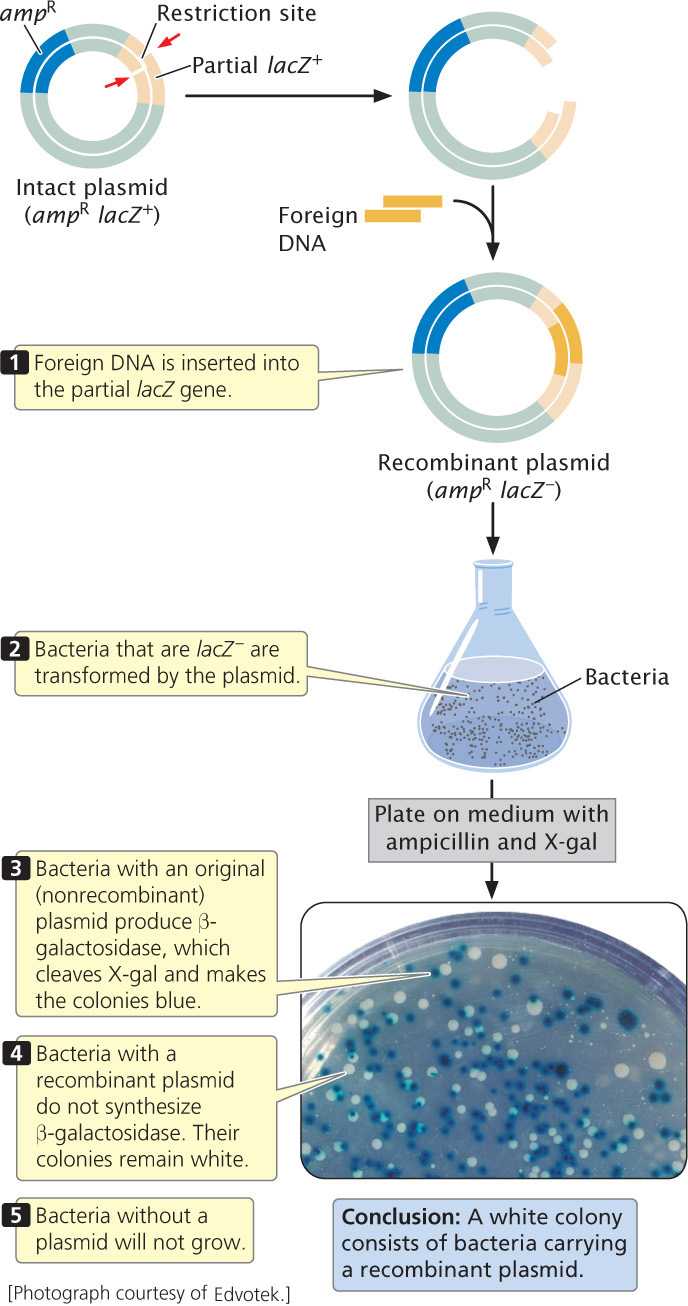
Bacteria that are lacZ– are transformed by the plasmids and plated on medium that contains ampicillin. Only cells that have been successfully transformed and contain a plasmid with the ampicillin-resistance gene will survive and grow. Some of these cells will contain an intact plasmid, whereas others possess a recombinant plasmid. The medium also contains the chemical X-gal, which produces a blue substance when cleaved. Bacterial cells with an intact original plasmid—without an inserted fragment—have a functional lacZ gene (the front end of the gene provided by the plasmid and the back end provided by the bacteria). These bacteria can synthesize β-galactosidase, which cleaves X-gal and turns the bacteria blue. Bacterial cells with a recombinant plasmid, however, have the front end of the β-galactosidase gene disrupted by the inserted DNA; they do not synthesize β-galactosidase and remain white. (In these experiments, the bacterium’s own β-galactosidase gene has been inactivated, and so only bacteria with the plasmid turn blue.) Thus, the color of the bacterial colony allows quick determination of whether a recombinant or intact plasmid is present in the cell. After cells with the recombinant plasmid have been identified, they can be grown in large numbers, replicating the inserted fragment of DNA.
Plasmids make ideal cloning vectors but can hold only DNA less than about 15 kb in size. When large DNA fragments are inserted into a plasmid vector, the plasmid becomes unstable.
Other Gene Vectors
A number of other vectors have been developed for cloning larger pieces of DNA in bacteria. For example, bacteriophage λ, which infects E. coli, can be used to clone up to 23,000 bp of foreign DNA; it transfers DNA into bacterial cells with high efficiency. Cosmids are plasmids that are packaged into empty viral protein coats and transferred to bacteria by viral infection. They can carry more than twice as much foreign DNA as can a phage vector. Bacterial artificial chromosomes (BACs) are vectors originally constructed from the F plasmid (a special plasmid that controls mating and the transfer of genetic material in some bacteria; see Chapter 9) and can hold very large fragments of DNA that can be as long as 300,000 bp. Table 19.2 compares the properties of plasmids, phage λ vectors, cosmids, and BACs.
544
| Cloning Vector | Size of DNA That Can Be Cloned | Method of Propagation | Introduction to Bacteria |
|---|---|---|---|
| Plasmid | As large as 15 kb | Plasmid replication | Transformation |
| Phage lambda | As large as 23 kb | Phage reproduction | Phage infection |
| Cosmid | As large as 44 kb | Plasmid reproduction | Phage infection |
| Bacterial artificial chromosome | As large as 300 kb | Plasmid reproduction | Electroporation |
| Note: 1 kb = 1000 bp. Electroporation consists of electrical pulses that increase the permeability of a membrane. | |||
Sometimes the goal in gene cloning is not just to replicate the gene, but also to produce the protein that it encodes. To ensure transcription and translation, a foreign gene is usually inserted into an expression vector, which, in addition to the usual origin of replication, restriction sites, and selectable markers, contains sequences required for transcription and translation in bacterial cells (Figure 19.9).
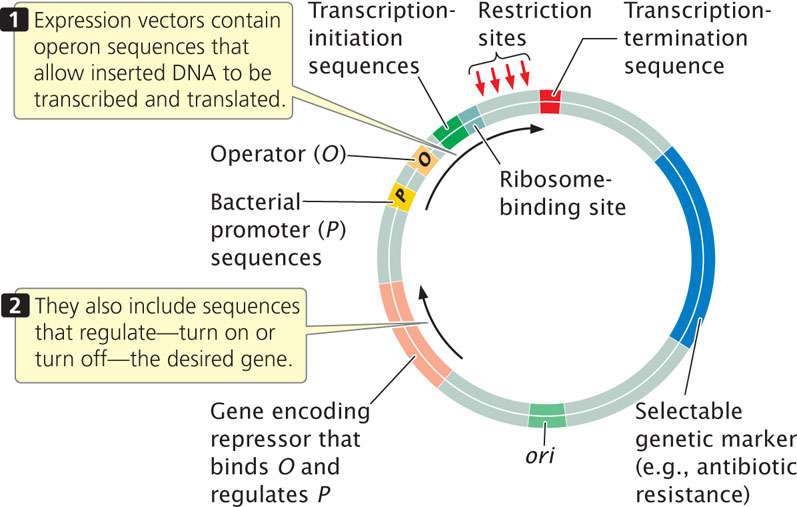
Although manipulating genes in bacteria is simple and efficient, the goal may be to transfer a gene into eukaryotic cells. For example, it might be desirable to transfer a gene conferring herbicide resistance into a crop plant, or to transfer a gene for clotting factor into a person suffering from hemophilia. Many eukaryotic proteins are modified after translation (e.g., sugar groups may be added). Such modifications are essential for proper function, but bacteria do not have the capacity to carry out the modification; thus a functional protein can be produced only in a eukaryotic cell.
A number of cloning vectors have been developed that allow the insertion of genes into eukaryotic cells. Special plasmids have been developed for cloning in yeast, and retroviral vectors have been developed for cloning in mammals. A yeast artificial chromosome (YAC) is a DNA molecule that has a yeast origin of replication, a pair of telomeres, and a centromere. These features ensure that YACs are stable, replicate, and segregate in the same way as yeast chromosomes. YACs are particularly useful because they can carry DNA fragments as large as 600 kb, and some special YACs can carry inserts of more than 1000 kb. YACs have been modified so that they can be used in eukaryotic organisms other than yeast.
The soil bacterium Agrobacterium tumefaciens, which invades plants through wounds and induces crown galls (tumors), has been used to transfer genes to plants. This bacterium contains a large plasmid called the Ti plasmid, part of which is transferred to a plant cell when A. tumefaciens infects a plant. In the plant, part of the Ti plasmid DNA integrates into one of the plant chromosomes, where it is transcribed and translated to produce several enzymes that help support the bacterium (Figure 19.10a).
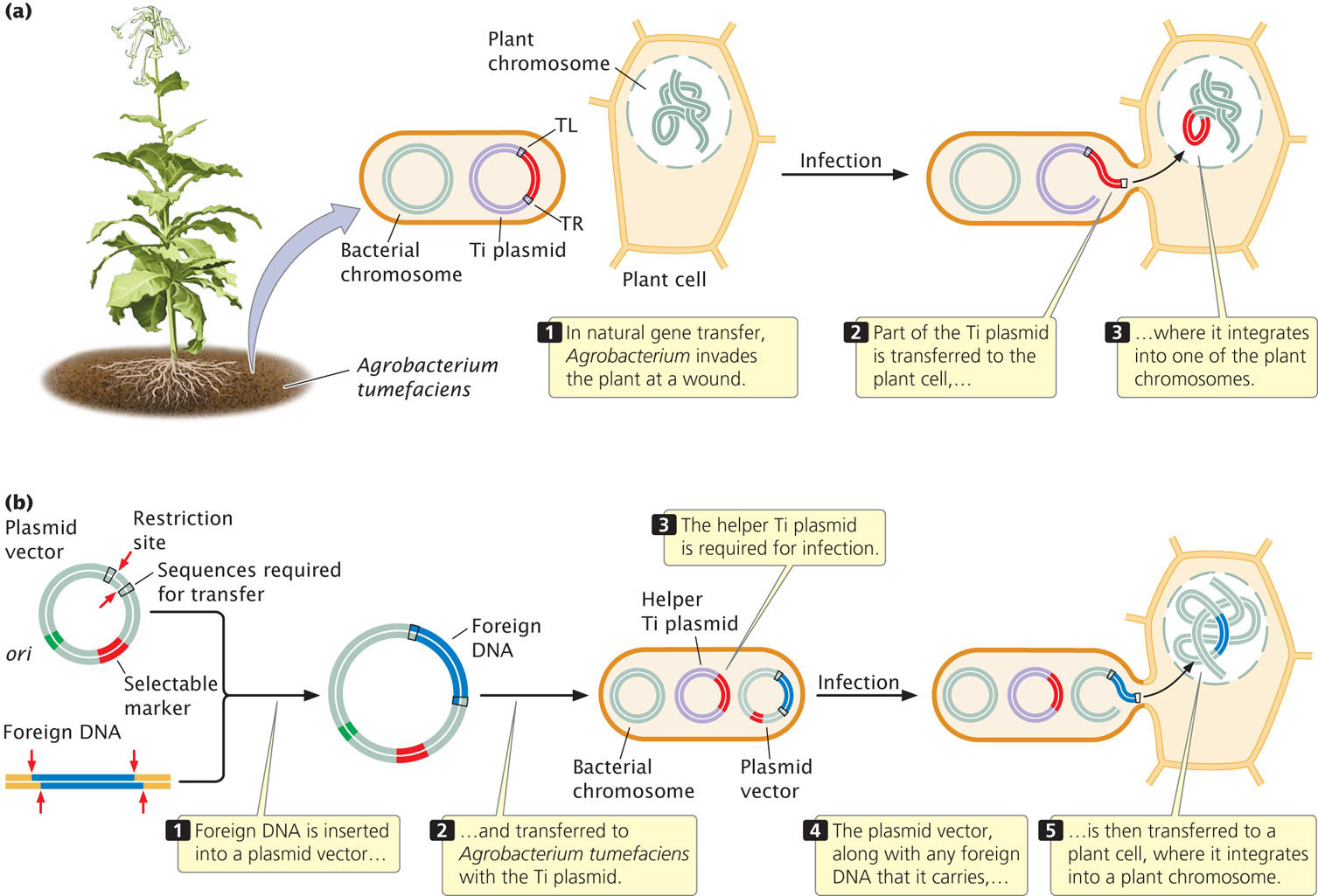
Geneticists have engineered a vector that contains the flanking sequences required to transfer DNA (called TL and TR, see Figure 19.10a), a selectable marker, and restriction sites into which foreign DNA can be inserted (Figure 19.10b). When placed in A. tumefaciens with a helper Ti plasmid (which carries sequences allowing the bacterium to infect the plant cell, which the artificial vector lacks), this vector will transfer the foreign DNA that it carries into a plant cell, where it will integrate into a plant chromosome. This vector has been used to transfer genes that confer economically significant attributes such as resistances to herbicides, plant viruses, and insect pests. ![]() TRY PROBLEM 31
TRY PROBLEM 31
CONCEPTS
DNA fragments can be inserted into cloning vectors, stable pieces of DNA that will replicate within a cell. A cloning vector must have an origin of replication, one or more unique restriction sites, and selectable markers. An expression vector contains sequences that allow a cloned gene to be transcribed and translated. Special cloning vectors have been developed for introducing genes into eukaryotic cells.
CONCEPT CHECK 4
How is a gene inserted into a plasmid cloning vector?
545
Application: The Genetic Engineering of Plants with Pesticides
An early and economically important use of molecular techniques for transferring genes to other organisms is the genetic engineering of plants that produce their own insecticides. The bacterium Bacillus thuringiensis naturally produces a protein, known as the Bt toxin, that is lethal to many insects. The Bt toxin is particularly attractive as an insecticide because it is specific to particular insects, breaks down quickly in the environment, and is not toxic to humans and other animals.
In 1987, Mark Vaeck and his colleagues at Plant Genetic Systems in Belgium genetically engineered tobacco plants to express the Bt toxin. Other researchers had isolated the gene that encodes the Bt toxin from B. thuringiensis and cloned it into a plasmid of E. coli so that the gene sequences could be easily manipulated (Figure 19.11). Vaeck and his colleagues used restriction enzymes to cleave the Bt gene sequences from these plasmids into fragments of different sizes. The Bt gene fragments were attached to a gene (neo) that confers resistance to the antibiotic kanamycin, which is toxic to plants and other eukaryotes, providing a selectable marker for cells containing the Bt gene. These synthetic sequences (called genetic constructs) contained fragments of the Bt gene of different lengths linked to a neo gene (see Figure 19.11). The constructs were inserted into an expression vector that contained a promoter, to ensure that the introduced sequences would be transcribed. The expression vector also contained poly(A) consensus sequences, which ensured that the mRNA produced from the fused Bt and neo genes would be processed properly and translated in the plant cells.
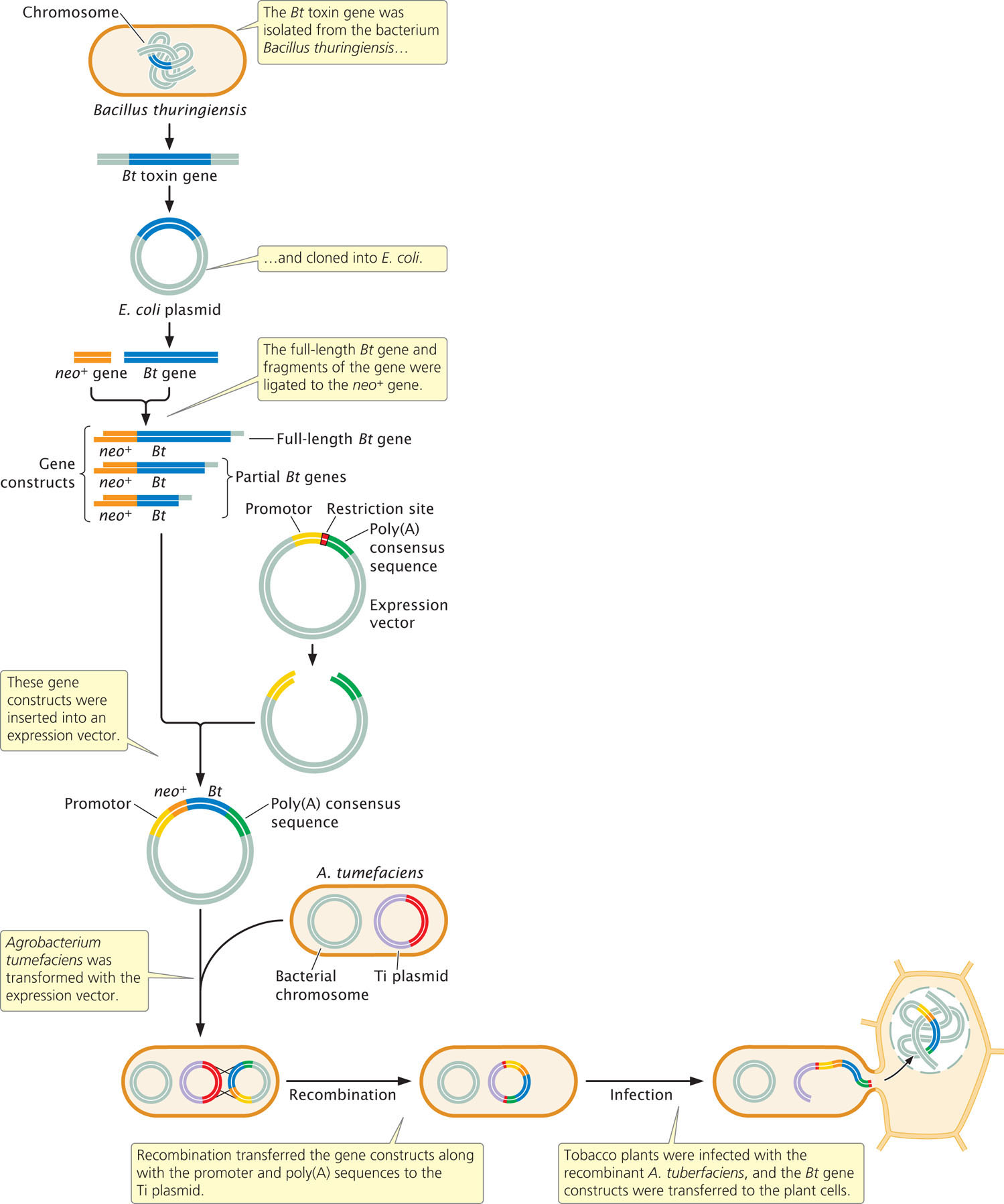
Agrobacterium tumefaciens bacteria were then transformed with the expression vectors. After the vectors were inside the Agrobacterium, sequences on the vector recombined with sequences on a Ti plasmid, transferring the gene constructs to the Ti plasmid. Small discs of leaves from a tobacco plant were infected with the genetically engineered Agrobacterium, which transferred the Ti plasmids into the plant cells. Whole tobacco plants were regenerated from the leaf disks and selected for resistance to kanamycin.
546
The resulting transgenic plants were then tested for resistance to insect pests. Leaves of the plants were fed to tobacco hornworms, and mortality rates of the hornworms were monitored. High mortality of hornworms was observed in about two-thirds of the plants containing fragments of the Bt gene. Interestingly, none of the plants containing the full-length Bt gene produced any insect-killing toxins; apparently, the plant cells were better able to translate the fragments of the Bt gene than to translate the entire gene. The transgenic plants producing Bt toxin grew normally. They were interbred to produce F1 plants, which also exhibited insect and kanamycin resistance, indicating that the introduced genes were stably integrated into the plant chromosomes.
Following the research done by Vaeck and his colleagues, other researchers used similar methods to introduce the gene for Bt toxin into cotton, tomatoes, corn, and other plants (Figure 19.12). Today, transgenic plants expressing the Bt toxin are used broadly throughout the world.

Amplifying DNA Fragments with the Polymerase Chain Reaction
The manipulation and analysis of genes require multiple copies of the DNA sequences used. A major problem in working at the molecular level is that each gene is a tiny fraction of the total cellular DNA. Because each gene is rare, it must be isolated and amplified before it can be studied. One way to amplify a DNA fragment is to clone it in bacterial cells. Indeed, for many years, gene cloning was the only way to amplify a DNA fragment, and cloning was a prerequisite for many other molecular methods. However, cloning is labor intensive and requires at least several days to grow the bacteria. Today, the polymerase chain reaction makes the amplification of short DNA fragments possible without cloning, although cloning is still widely used for amplifying large DNA fragments and for other manipulations of DNA sequences.
The polymerase chain reaction (PCR), first developed in 1983 by Kary Mullis, allows DNA fragments to be amplified a billionfold within just a few hours. It can be used with extremely small amounts of original DNA, even a single molecule. The polymerase chain reaction has revolutionized molecular biology and is now one of the most widely used molecular techniques. The basis of PCR is replication catalyzed by a DNA polymerase. Replication in this case has two essential requirements: (1) a DNA template from which a new DNA strand can be copied and (2) a pair of primers with a 3′-OH group to which new nucleotides can be added.
547
Because a DNA molecule consists of two nucleotide strands, each of which can serve as a template, the amount of DNA doubles with each replication event. The primers used in PCR to replicate the templates are short fragments of DNA, typically from 17 to 25 nucleotides long, that are complementary to known sequences on the template.
The Polymerase Chain Reaction
To carry out PCR, researchers begin with a solution that includes the target DNA, DNA polymerase, all four deoxyribonucleoside triphosphates (dNTPs—the substrates for DNA polymerase), primers, and magnesium ions and other salts that are necessary for the reaction to proceed. A typical polymerase chain reaction includes three steps (Figure 19.13). In step 1, a starting solution of DNA is heated to between 90° and 100°C to break the hydrogen bonds between the strands and thus produce the necessary single-stranded templates. The reaction mixture is held at this temperature for only a minute or two. In step 2, the DNA solution is cooled quickly to between 30° and 65°C and held at this temperature for a minute or less. During this short interval, the DNA strands will not have a chance to reanneal, but the primers will be able to attach to the template strands. In step 3, the solution is heated for a minute or less to 72°C, the temperature at which DNA polymerase can synthesize new DNA strands. At the end of the cycle, two new double-stranded DNA molecules are produced for each original molecule of target DNA.
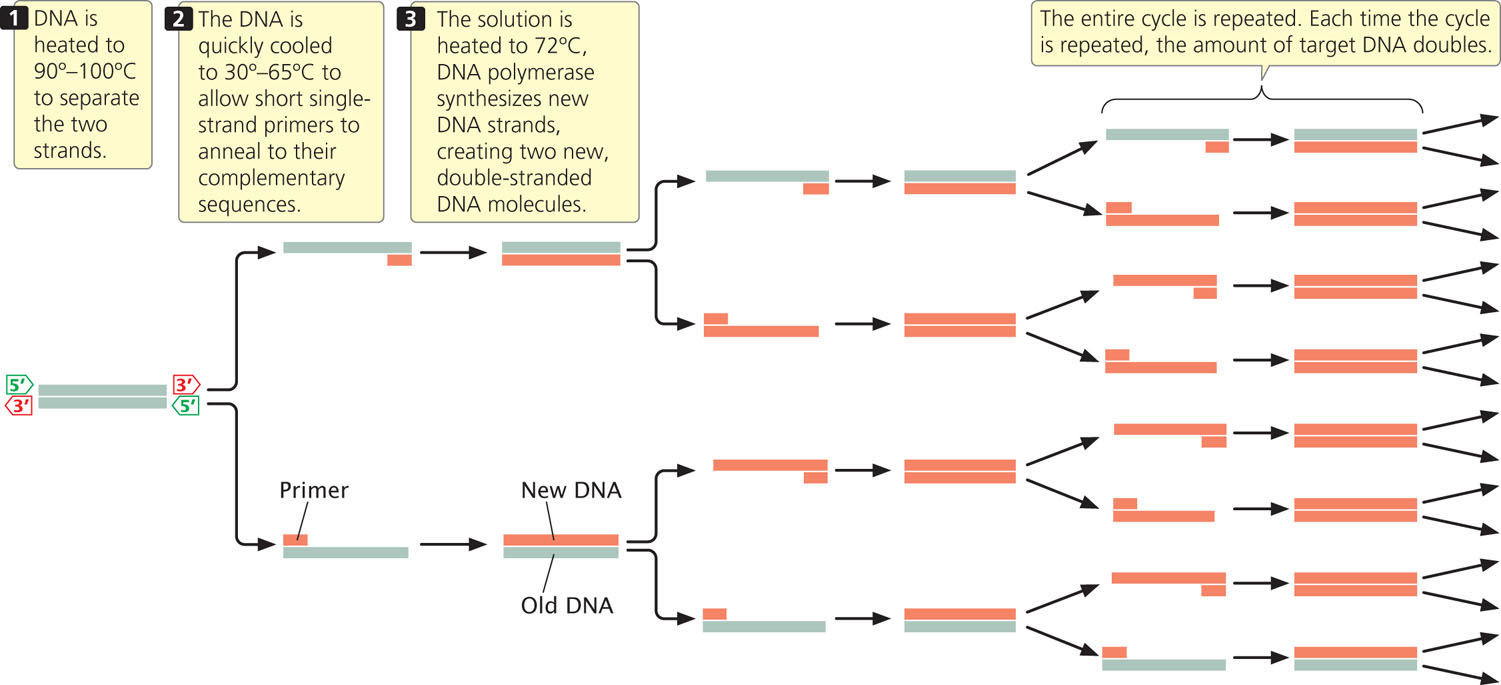
The whole cycle is then repeated. With each cycle, the amount of target DNA doubles; so the amount of target DNA increases geometrically. One molecule of DNA increases to more than 1000 molecules in 10 PCR cycles, to more than 1 million molecules in 20 cycles, and to more than 1 billion molecules in 30 cycles. Each cycle is completed within a few minutes, so a large amplification of DNA can be achieved within a few hours. To see how the polymerase chain reaction quickly increases the number of copies of a DNA fragment, view Animation 19.2.
548
Two key innovations facilitated the use of PCR in the laboratory. The first was the discovery of a DNA polymerase that is stable at the high temperatures used in step 1 of PCR. The DNA polymerase from E. coli that was originally used in PCR denatures at 90°C. For this reason, fresh enzyme had to be added to the reaction mixture in each cycle, slowing the process considerably. This obstacle was overcome when DNA polymerase was isolated from the bacterium Thermus aquaticus, which lives in the boiling springs of Yellowstone National Park. This enzyme, dubbed Taq polymerase, is remarkably stable at high temperatures and is not denatured in the strand-separation step of PCR; it can be added to the reaction mixture at the beginning of the PCR process and will continue to function through many cycles.
The second key innovation was the development of automated thermal cyclers—machines that bring about the rapid temperature changes necessary for the different steps of PCR. Originally, tubes containing reaction mixtures were moved by hand among water baths set at the different temperatures required for the three steps of each cycle. In automated thermal cyclers, the reaction tubes are placed in a metal block that changes temperature rapidly according to a computer program.
In addition to amplifying DNA, PCR can be used to amplify sequences corresponding to RNA. This amplification is accomplished by first converting RNA into complementary DNA (cDNA) with the use of the enzyme reverse transcriptase. The cDNA can then be amplified by the usual PCR. This technique is referred to as reverse-transcription PCR.
Limitations of PCR
The polymerase chain reaction is now often used in place of gene cloning, but it has several limitations. First, the use of PCR requires prior knowledge of at least part of the sequence of the target DNA to allow the construction of the primers. Second, the capacity of PCR to amplify extremely small amounts of DNA makes contamination a significant problem. Minute amounts of DNA from the skin of laboratory workers or small particles in the air can enter a reaction tube and be amplified along with the target DNA. Careful laboratory technique and the use of controls are necessary to circumvent this problem.
A third limitation of PCR is accuracy. Unlike other DNA polymerases, Taq polymerase does not have the capacity to proofread and, under standard PCR conditions, it incorporates an incorrect nucleotide about once every 20,000 bp. New heat-stable DNA polymerases with proofreading capacity have been isolated, giving more accurate PCR results.
A fourth limitation of PCR is that the size of the fragments that can be amplified by standard Taq polymerase is usually less than 2000 bp. By using a combination of Taq polymerase and a DNA polymerase with proofreading capacity, and by modifying the reaction conditions, investigators have been successful in extending PCR amplification to larger fragments, but even these larger fragments are limited in length to 50,000 bp or smaller.
Applications of PCR
In spite of its limitations, PCR has become one of the most widely used tools within molecular biology. The primers used in PCR are specific for known DNA sequences, and so PCR can be used to detect the presence of a particular DNA sequence in a sample. For example, PCR is often used to detect the presence of viruses in blood samples by adding primers complementary to known viral DNA sequences to the reaction. If viral DNA is present, the primers will attach to it, and PCR will amplify a DNA fragment of known length. The presence of a DNA fragment of the proper length on a gel indicates the presence of viral DNA in the blood sample. Modern diagnostic tests for infection with HIV, the causative agent of AIDS (see Chapter 9), use this type of PCR amplification of HIV sequences.
Another common application of PCR is to identify genetic variation in natural populations. PCR can be used to amplify specific segments of DNA, which are then analyzed with other molecular tools that identify differences in nucleotide sequences. Also, primers specific to a particular DNA variant can be used to determine whether that variant is present in the genome of an individual organism.
PCR has also allowed the isolation of DNA from ancient sources, such as DNA from Neanderthals (discussed in the introduction to Chapter 10). It is commonly used to amplify small amounts of DNA from crime scenes and identify the source of a DNA sample through detection of microsatellite variation (see section on DNA fingerprinting later in this chapter).
PCR can be used to introduce new sequences into a fragment of DNA. Although the 3′ end of the primers used in PCR must be complementary to the template, there can be flexibility in the sequences at the 5′ end of the primer. Primers can be designed so that they contain new restriction sites or other desirable sequences at their 5′ ends. These sequences will then be copied by the reaction and thus added to the ends of the DNA amplified by PCR.
A modification of PCR, known as real-time PCR, can be used to quantitatively determine the amount of starting nucleic acid. In this procedure, normal PCR is used to amplify a specific DNA fragment, and a sensitive instrument is used to accurately determine the amount of DNA that is present in solution after each PCR cycle. A probe that fluoresces and is specific to the DNA sequence of interest is frequently used in the reaction, and so only the DNA of interest is measured. The technique is called real-time PCR because the amount of DNA amplified is measured as the reaction proceeds.
Often, real-time PCR is combined with reverse-transcription PCR to measure the amount of mRNA in a sample, allowing biologists to determine the level of gene expression in different cells and under different conditions. For example, researchers interested in how gene expression changes in response to the administration of a drug often use real-time PCR to quantitatively measure the amount of mRNA produced by specific genes in cells exposed to the drug and compare it with the amount of mRNA produced by the genes in control cells with no drug exposure.
549
CONCEPTS
The polymerase chain reaction is an enzymatic, in vitro method for rapidly amplifying DNA. In this process, DNA is heated to separate the two strands, short primers attach to the target DNA, and DNA polymerase synthesizes new DNA strands from the primers. Each cycle of PCR doubles the amount of DNA. PCR has a number of important applications in molecular biology.
CONCEPT CHECK 5
Why is the use of a heat-stable DNA polymerase important to the success of PCR?