19.5 Molecular Techniques Are Increasingly Used to Analyze Gene Function
In the preceding sections, we learned about powerful molecular techniques for isolating, recombining, and analyzing DNA sequences. Although these techniques provide a great deal of information about the organization and nature of gene sequences, the ultimate goal of many molecular studies is to better understand the function of the sequences. In this section, we will explore some advanced molecular techniques that are frequently used to determine gene function and to better understand the genetic processes that these sequences undergo.
Forward and Reverse Genetics
The traditional approach to the study of gene function begins with the isolation of mutant organisms. For example, suppose a geneticist is interested in genes that affect cardiac function in mammals. A first step would be to find individuals—perhaps mice—that have hereditary defects in heart function. The mutations causing the cardiac problems in the mice could then be mapped, and the implicated genes could be isolated and sequenced. The proteins produced by the genes could then be predicted from the gene sequences and isolated. Finally, the biochemistry of the proteins could be studied and their role in heart function discerned. This approach, which begins with a phenotype (a mutant individual) and proceeds to a gene that encodes the phenotype, is called forward genetics.
564
An alternative approach is to begin with a genotype—a DNA sequence—and proceed to the phenotype by altering the sequence or inhibiting its expression. A geneticist might begin with a gene of unknown function, induce mutations in it, and then look to see what effect these mutations have on the phenotype of the organism. This approach is called reverse genetics. Today, both forward and reverse genetic approaches are widely used in analyses of gene function.
Creating Random Mutations
Forward genetics depends on the identification and isolation of random mutations that affect a phenotype of interest. Early in the study of genetics, geneticists were forced to rely on naturally occurring mutations, which are usually rare and can be detected only if large numbers of organisms are examined. The discovery of mutagenic agents—environmental factors that increase the rate of mutation (see Chapter 18)—provided a means of increasing the number of mutants in experimental populations of organisms. One of the first examples of experimentally created mutations was Hermann Muller’s use of X-rays in 1927 to induce X-linked mutations in Drosophila melanogaster.
Today, radiation, chemical mutagens, and transposable elements are all used to create mutations for genetic analysis. To determine all genes that might affect a phenotype, it is desirable to create mutations in as many genes as possible—that is, to saturate the genome with mutations. This procedure is done with a mutagenic screen, which will be described in Chapter 20.
Site-Directed Mutagenesis
Reverse genetics depends on the ability to create mutations, not at random, but in particular DNA sequences, and then to study the effects of these mutations on the organism. Mutations are induced at specific locations through a process called site-directed mutagenesis.
A number of different strategies have been developed for site-directed mutagenesis. One strategy that is often used in bacteria is to cut out a short sequence of nucleotides with restriction enzymes and replace it with a short, synthetic oligonucleotide that contains the desired mutated sequence. The success of this method depends on the availability of restriction sites flanking the sequence to be altered.
If appropriate restriction sites are not available, oligonucleotide-directed mutagenesis can be used (Figure 19.32). In this method, a single-stranded oligonucleotide is produced that differs from the target sequence by one or a few bases. Because they differ in only a few bases, the target DNA and the oligonucleotide will pair. When successfully paired with the target DNA, the oligonucleotide can act as a primer to initiate DNA synthesis, which produces a double-stranded molecule with a mismatch in the primer region. When this DNA is transferred to bacterial cells, the mismatched bases will be repaired by bacterial enzymes. About half of the time the normal bases will be changed into mutant bases, and about half of the time the mutant bases will be changed into normal bases. The bacteria are then screened for the presence of the mutant gene.
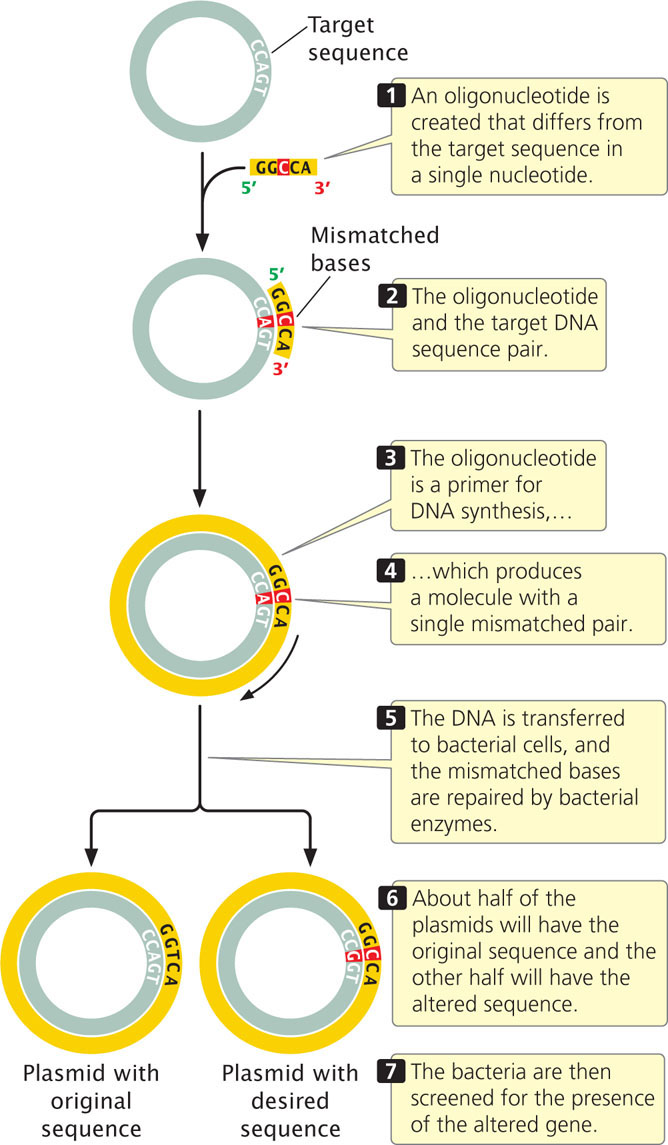
565
CONCEPTS
Forward genetics begins with a phenotype and detects and analyzes the genotype that causes the phenotype. Reverse genetics begins with a gene sequence and, through analysis, determines the phenotype that it encodes. Particular mutations can be introduced at specific sites within a gene by means of site-directed and oligonucleotide-directed mutagenesis.
 CONCEPT CHECK 10
CONCEPT CHECK 10
A geneticist interested in immune function induces random mutations in a number of specific genes in mice and then determines which of the resulting mutant mice have impaired immune function. This procedure is an example of
- forward genetics.
- reverse genetics.
- both forward and reverse genetics.
- neither forward nor reverse genetics.
Transgenic Animals
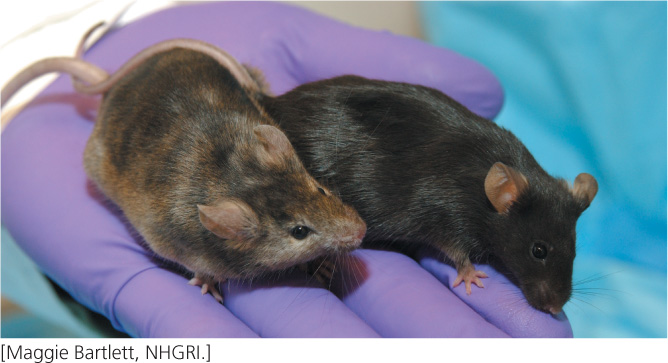
Another way that gene function can be analyzed is by adding DNA sequences of interest to the genome of an organism that normally lacks such sequences and then seeing the effect that the introduced sequence has on the organism’s phenotype. This method is a form of reverse genetics. An organism that has been permanently altered by the addition of a DNA sequence to its genome is said to be transgenic, and the foreign DNA that it carries is called a transgene (Figure 19.33). Here, we consider techniques for the creation of transgenic mice, which are often used in the study of the function of human genes because they can be genetically manipulated in ways that are impossible with humans and, as mammals, they are more similar to humans than are fruit flies, fish, and other model genetic organisms.
The oocytes of mice and other mammals are large enough that DNA can be injected into them directly. Immediately after penetration by a sperm, a fertilized mouse egg contains two pronuclei, one from the sperm and one from the egg; these pronuclei later fuse to form the nucleus of the embryo. Mechanical devices can manipulate extremely fine, hollow glass needles to inject DNA directly into one of the pronuclei of a fertilized egg (Figure 19.34). Typically, a few hundred copies of cloned, linear DNA are injected into a pronucleus, and, in a few of the injected eggs, copies of the cloned DNA integrate randomly into one of the chromosomes through a process called nonhomologous recombination. After injection, the embryos are implanted in a pseudopregnant female—a surrogate mother that has been physiologically prepared for pregnancy by mating with a vasectomized male.
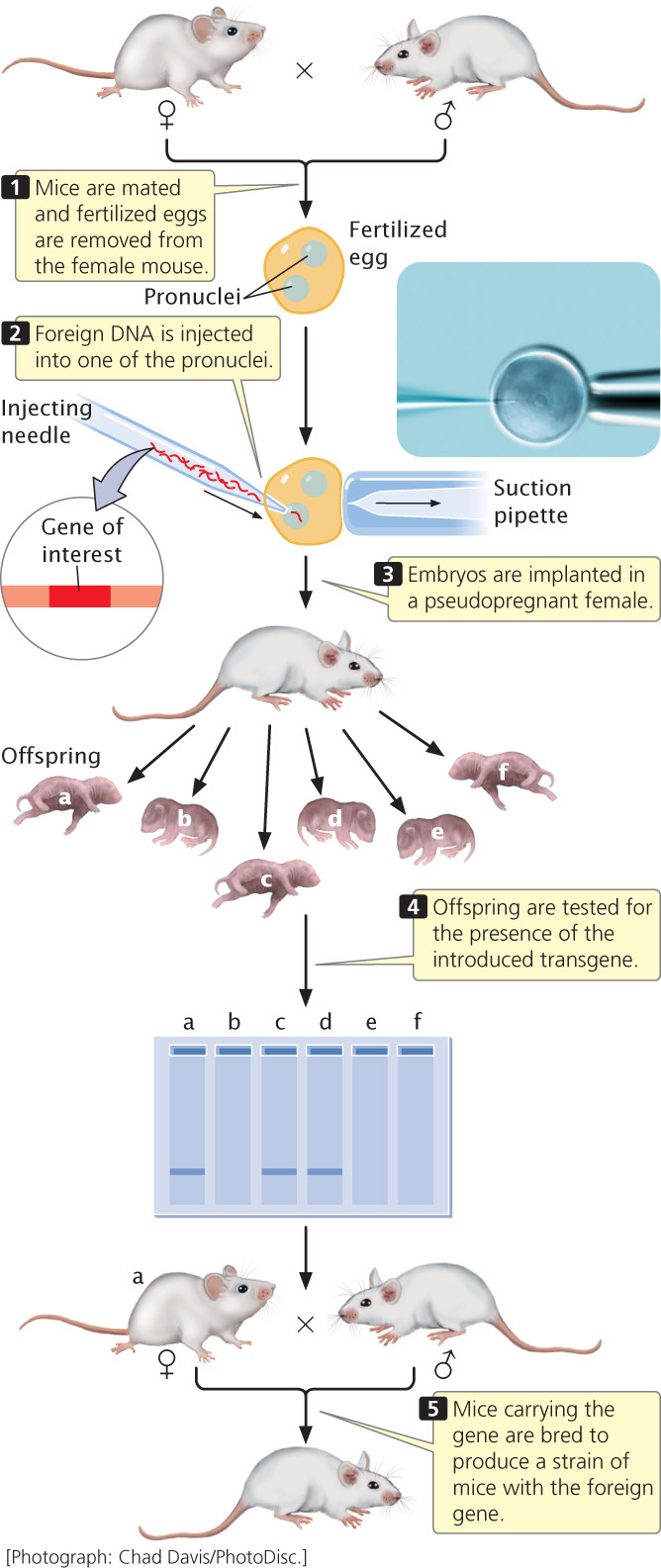
566
Only about 10% to 30% of the embryos survive and, of those that do survive, only a few have a copy of the cloned DNA stably integrated into a chromosome. Nevertheless, if several hundred embryos are injected and implanted, there is a good chance that one or more mice whose chromosomes contain the foreign DNA will be born. Moreover, because the DNA was injected at the one-cell stage of the embryo, these mice usually carry the cloned DNA in every cell of their bodies, including their reproductive cells, and will therefore pass the foreign DNA on to their progeny. Through interbreeding, a strain of mice that carry the foreign gene can be created.
Transgenic mice have proved useful in the study of gene function. For example, proof that the SRY gene (see Chapter 4) is the male-determining gene in mice was obtained by injecting a copy of the SRY gene into XX embryos and observing that these mice developed as males. In addition, researchers have created a number of transgenic mouse strains that serve as experimental models for human genetic diseases.
Knockout Mice
A useful variant of the transgenic approach is to produce mice in which a normal gene has been not just mutated, but fully disabled. These animals, called knockout mice, are particularly helpful in determining the function of a gene: the phenotype of the knockout mouse often gives a good indication of the function of the missing gene.
The creation of knockout mice begins when a normal gene is cloned in bacteria and then “knocked out,” or disabled. There are a number of ways to disable a gene, but a common method is to insert a gene called neo, which confers resistance to the antibiotic G418, into the middle of the target gene (Figure 19.35). The insertion of neo both disrupts the target gene and provides a convenient marker for finding copies of the disabled gene. In addition, a second gene, usually the herpes simplex viral thymidine kinase (tk) gene, is linked to the disrupted gene. The disabled gene is then transferred to cultured embryonic mouse cells, where it may exchange places with the normal copy on the mouse chromosome through homologous recombination.
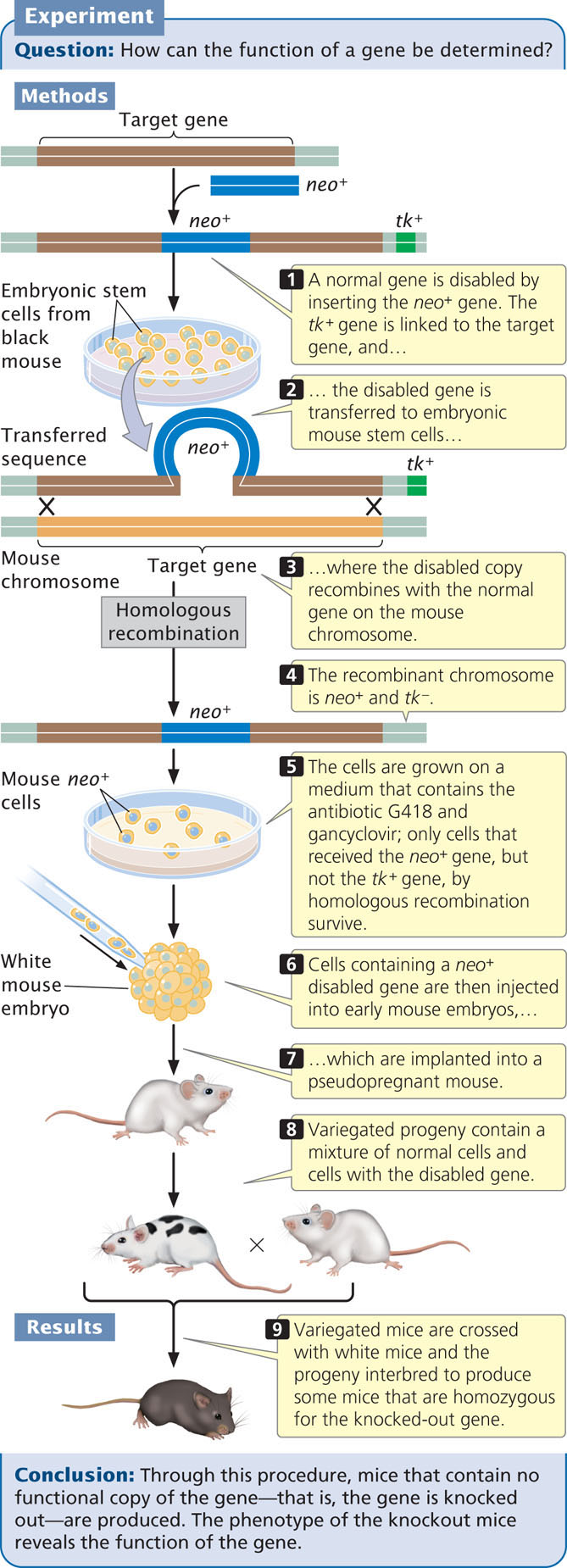
567
After the disabled gene has been transferred to the embryonic cells, the cells are screened by adding the antibiotic G418 to the medium. Only cells with the disabled gene containing the neo insert will survive. Because the frequency of nonhomologous recombination is higher than that of homologous recombination and because the intact target gene is replaced by the disabled copy only through homologous recombination, a means to select for the rarer homologous recombinants is required. The presence of the viral tk gene makes the cells sensitive to gancyclovir. Thus, transfected cells that grow on medium containing G418 and gancyclovir will contain the neo gene (disabled target gene) but not the adjacent tk gene, because the tk gene will be eliminated in the double-crossover event. These cells contain the desired homologous recombinants. The nonhomologous recombinants (random insertions) will contain both the neo and the tk genes, and these transfected cells will die on the selection medium owing to the presence of gancyclovir. The surviving cells are injected into an early-stage mouse embryo, which is then implanted into a pseudopregnant mouse. Cells in the embryo carrying the disabled gene and normal embryonic cells carrying the wild-type gene will develop together, producing a chimera—a mouse that is a genetic mixture of the two cell types. The production of chimeric mice is not itself desirable, but replacing all the cells of the embryo with injected cells is impossible.
Chimeric mice can be easily identified if the injected embryonic cells came from a black mouse and the embryos into which they are injected came from a white mouse; the resulting chimeras will have variegated black and white fur. Some of the chimeras may have the knockout gene in their germline cells and can transmit it to the next generation. The chimeras are crossed to white mice; any black progeny are heterozygous for the knockout. The black progeny can then be intercrossed to produce some progeny that are homozygous for the knockout gene. The effects of disabling a particular gene can be observed in these homozygous mice. Three scientists who helped develop the techniques for creating knockout mice—Mario Capecchi, Oliver Smithies, and Martin Evans—were awarded the Nobel Prize in physiology or medicine in 2007 for their work.
Additional techniques have been developed for knocking out genes only in certain tissues or at certain times; these are called conditional knockouts. A variant of the knockout procedure is to insert in mice a particular DNA sequence into a known chromosome location. For example, researchers might insert the sequence of a human disease-causing allele into the same locus in mice, creating a precise mouse model of the human disease. Mice that carry inserted sequences at specific locations are called knock-in mice.
CONCEPTS
A transgenic mouse is produced by the injection of cloned DNA into the pronucleus of a fertilized egg, followed by implantation of the egg into a female mouse. In knockout mice, the injected DNA contains a mutation that disables a gene. Inside the mouse embryo, the disabled copy of the gene can exchange with the normal copy of the gene through homologous recombination.
CONCEPT CHECK 11
What is the advantage of using the neo gene to disrupt the function of a gene in knockout mice?
- The neo gene produces an antibiotic that kills unwanted cells.
- The neo gene is the right size for disabling other genes.
- The neo gene provides a selectable marker for finding cells that contain the disabled gene.
- The neo gene produces a toxin that inhibits transcription of the target gene.
Silencing Genes with RNAi
In the preceding sections, we considered the analysis of gene function by introducing mutations or new DNA sequences into the genome and analyzing the resulting phenotype to provide information about the function of the altered or introduced DNA. We could also analyze gene function by temporarily turning a gene off and seeing what effect the absence of the gene product has on the phenotype. For many years, there was no method for selectively affecting gene expression. However, the discoveries of siRNAs (small interfering RNAs) and miRNAs (microRNAs; see Chapters 14 and 17) provided powerful tools for controlling the expression of individual genes.
Recall that siRNAs and miRNAs are small RNA molecules that combine with proteins to form the RNA-induced silencing complex (RISC). In a process called RNA interference or RNAi, the RISC pairs with complementary sequences on mRNA and either cleaves the mRNA or prevents the mRNA from being translated. Molecular geneticists have exploited this natural machinery for turning off the expression of specific genes. Studying the effect of silencing a gene with siRNA can often be a source of insight into the gene’s function.
The first step in using RNAi technology is to design the siRNAs such that they will be recognized and cleaved by Dicer (the protein that processes siRNAs; see Chapter 14). The complementary sequence must be unique to the target mRNA and not be found on other mRNAs so that the siRNA will not inhibit nontarget mRNAs. Computer programs are often used to design optimal siRNAs.
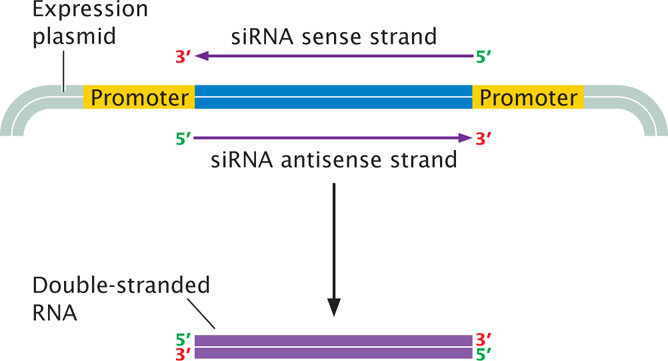
After the siRNA sequence has been designed, it must be synthesized. One way to create an siRNA is to use an oligonucleotide synthesizer to synthesize a DNA fragment corresponding to the siRNA sequence. The synthesized oligonucleotide can be cloned into a plasmid expression vector between two strong promoters (Figure 19.36). Escherichia coli are then transformed with the plasmid. Within the bacteria, transcription from the two promoters will proceed in both directions, producing two complementary RNA molecules that will pair to form a double-stranded RNA molecule recognized by Dicer. Alternatively, double-stranded RNA sequences can be synthesized directly with a gene synthesizer.
568
The next task is to deliver the double-stranded siRNA to the cells. Delivery can be done in a variety of ways, depending on the cell type. The model genetic organism Caenorhabditis elegans can be fed E. coli (their natural food) containing the expression vector. Transcription within the bacteria produces double-stranded RNA, that the worms ingest and incorporate into their cells. Alternatively, double-stranded siRNA can be injected directly into cells or the body cavity. Yet another approach is to synthesize a short sequence of DNA that has internal complementarity so that, when transcribed, it folds up into a short hairpin RNA (shRNA) with a double-stranded section. Within a cell, the shRNAs are processed by Dicer to produce siRNAs that bring about gene silencing. DNA sequences containing siRNAs can be introduced into a vector with the use of standard cloning techniques, and the vector can be used to deliver the DNA into a cell. An advantage of this approach is that, with the addition of a DNA sequence, the RNAi sequence has the potential to become a permanent part of the cell’s genome and be passed on to progeny.
One of the major advantages of siRNA for controlling gene expression is that it acts in trans—that is, a single copy of an siRNA gene will shut down expression of both copies of the target gene. Another advantage is that the target gene remains intact and therefore the silencing effects are reversible. ![]() TRY PROBLEM 43
TRY PROBLEM 43
Application: Using RNAi to Treat Human Disease
In addition to its value in determining gene function, RNAi holds potential as a therapeutic agent for the future treatment of human diseases. This potential includes using siRNAs against RNA viruses, such as HIV, as well as using them to treat genetic diseases and cancer.
Treatment of High Cholesterol With RNAi
Research has examined the potential of RNAi for the treatment of cholesterol metabolism disorders. Although cholesterol is essential for life, too much cholesterol is unhealthy: high blood cholesterol is a major contributor to heart disease, the leading cause of death in the United States. Cholesterol is normally transported throughout the body in the form of small particles called lipoproteins, which consist of a core of lipids surrounded by a shell of phospholipids and proteins (see Figure 6.6 in Chapter 6). The ApoB protein is an essential part of lipoproteins. Some people possess genetic mutations that cause elevated levels of ApoB, which predisposes them to coronary artery disease. Findings from studies suggest that lowering the amount of ApoB can reduce the number of lipoproteins and lower blood cholesterol in these people, as well as in people who have elevated cholesterol for other reasons.
In 2006, Tracy Zimmermann and her colleagues at Alnylam Pharmaceuticals and Protiva Biotherapeutics demonstrated that RNAi could be used to reduce the levels of ApoB and blood cholesterol in nonhuman primates. The investigators first created siRNAs that targeted apoB gene expression (apoB-siRNAs) on the basis of the known sequence of the gene. The apoB-siRNAs were synthesized in the laboratory and consisted of 21 nucleotides on the sense strand (the strand that was complementary to the apoB mRNA) and 23 nucleotides on the complementary antisense strand, with a two-nucleotide overhang.
The next task was to get the apoB-siRNA into the cell. Although siRNAs are readily taken up by the cells of invertebrates such as C. elegans, most siRNAs will not readily pass through the membranes of mammalian cells in a form that is still effective in gene silencing. In addition, siRNAs are rapidly removed from circulation. To overcome these problems, Zimmermann and her colleagues encapsulated the apoB-siRNAs in lipids, creating stable nucleic-acid–lipid particles (SNALPs). The SNALPs greatly increased the time spent by the siRNAs in circulation and enhanced their uptake by the cell.
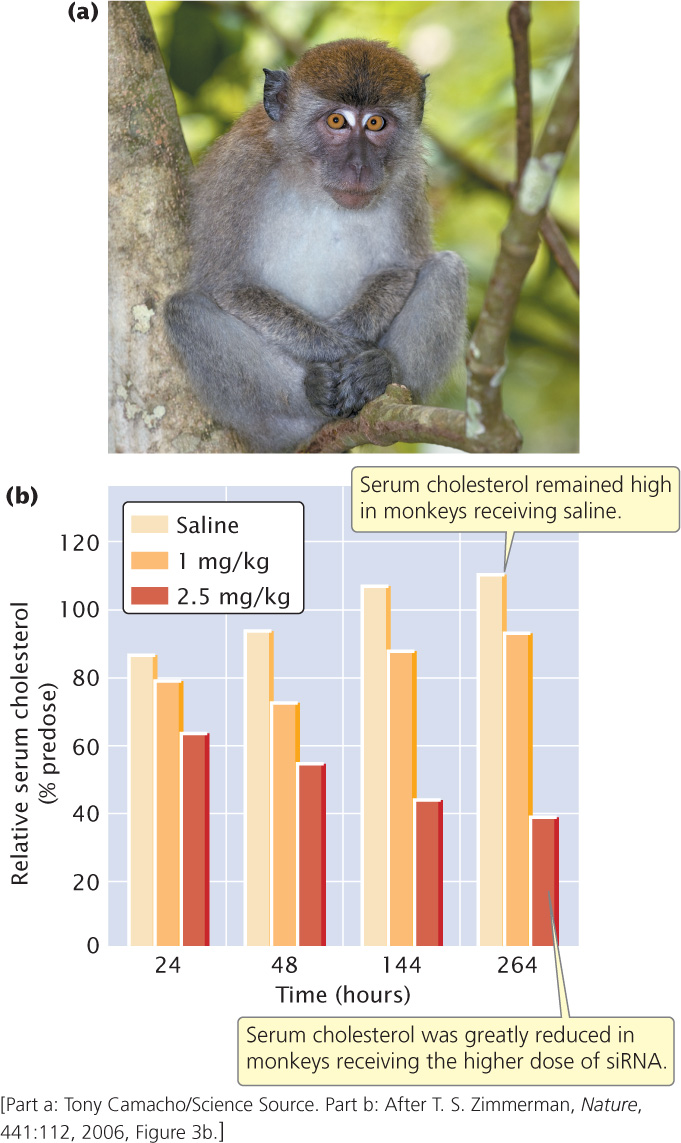
The researchers then tested the effects of the apoB-siRNAs on reducing the synthesis of the ApoB protein and on cholesterol levels. They injected cynomolgus monkeys (Figure 19.37a) with SNALPs containing apoB-siRNA at two doses: 1 mg/kg and 2.5 mg/kg. In addition, they injected a third group of monkeys with saline as a control. They found that the apoB-siRNA clearly silenced the apoB gene: 48 hours after treatment, apoB mRNA in the liver was reduced by 68% for monkeys receiving the 1-mg dose and 90% for monkeys receiving the 2.5-mg dose.
569
When the researchers examined serum cholesterol levels in the monkeys, they found that monkeys receiving the apoB-siRNA had a significant reduction in blood-cholesterol levels (Figure 19.37b). Importantly, they observed no negative effects of the siRNA treatment. Although preliminary, this study suggests that siRNAs have potential for future treatment of human diseases.