20.1 Structural Genomics Determines the DNA Sequences of Entire Genomes
Structural genomics concerns the organization and sequence of genetic information contained within a genome. Often, an early step in characterizing a genome is to prepare genetic and physical maps of its chromosomes. These maps provide information about the relative locations of genes, molecular markers, and chromosome segments, which are often essential for positioning chromosome segments and aligning stretches of sequenced DNA into a whole-genome sequence.
Genetic Maps
Everyone has used a map at one time or another. Maps are indispensable for finding a new friend’s house, the way to an unfamiliar city in your state, or the location of a country. Each of these examples requires a map with a different scale. To find a friend’s house, you would probably use a city street map; to find your way to an unknown city, you might pick up a state highway map; to find a country such as Kazakhstan, you would need a world atlas. Similarly, navigating a genome requires maps of different types and scales.
Construction of Genetic maps
Genetic maps (also called linkage maps) provide a rough approximation of the locations of genes relative to the locations of other known genes (Figure 20.1). These maps are based on the genetic function of recombination (hence the name genetic map). The basic principles of constructing genetic maps are discussed in detail in Chapter 7. In short, individual organisms of known genotype are crossed, and the frequency of recombination between loci is determined by examining the progeny. If the recombination frequency between two loci is 50%, then the loci are found on different chromosomes or are far apart on the same chromosome. If the recombination frequency is less than 50%, the loci are found close together on the same chromosome (they belong to the same linkage group). For linked genes, the rate of recombination is proportional to the physical distance between the loci. Distances on genetic maps are measured in percent recombination (centimorgans, cM), or map units (m.u.). Data from multiple two-point or three-point crosses can be integrated into linkage maps for whole chromosomes.
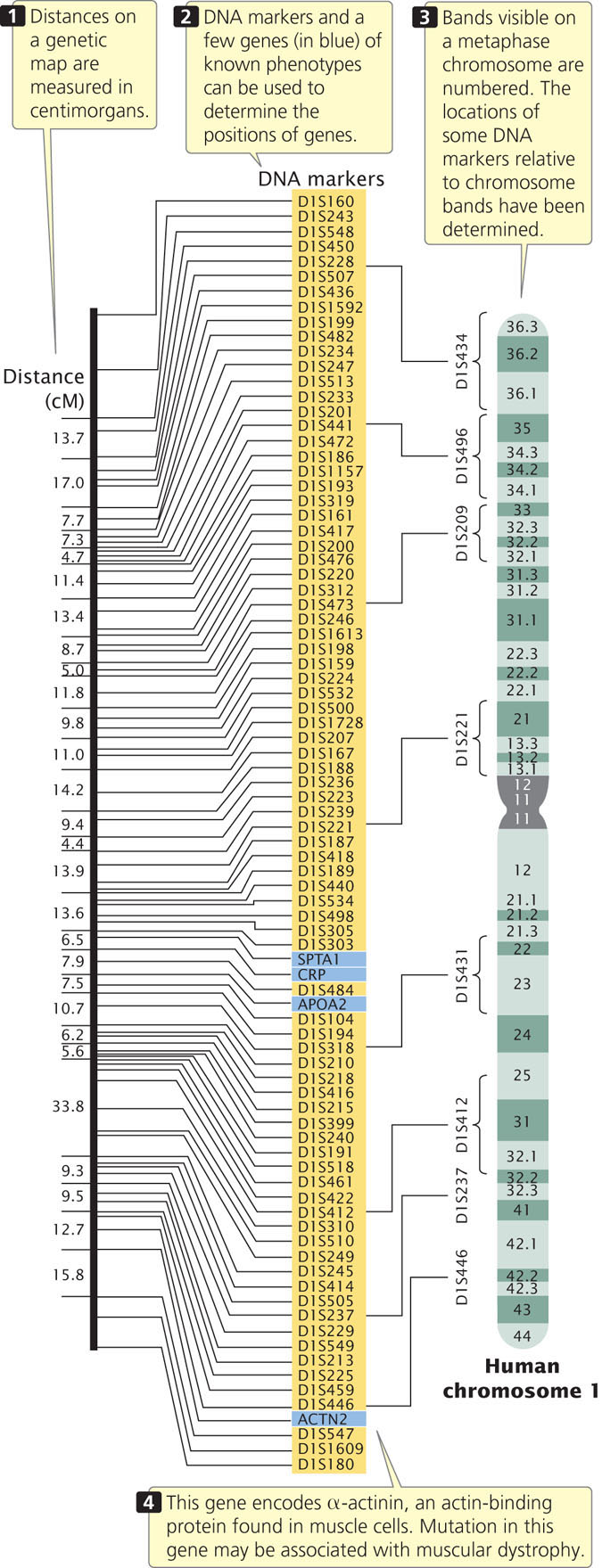
For many years, genes could be detected only by observing their influence on a trait (the phenotype), and the construction of genetic maps was limited by the availability of single-locus traits that could be examined for evidence of recombination. Eventually, this limitation was overcome by the development of molecular techniques, such as the analysis of restriction fragment length polymorphisms, the polymerase chain reaction, and DNA sequencing (see Chapter 19), which are able to provide molecular markers that can be used to construct and refine genetic maps.
Limitations of Genetic Maps
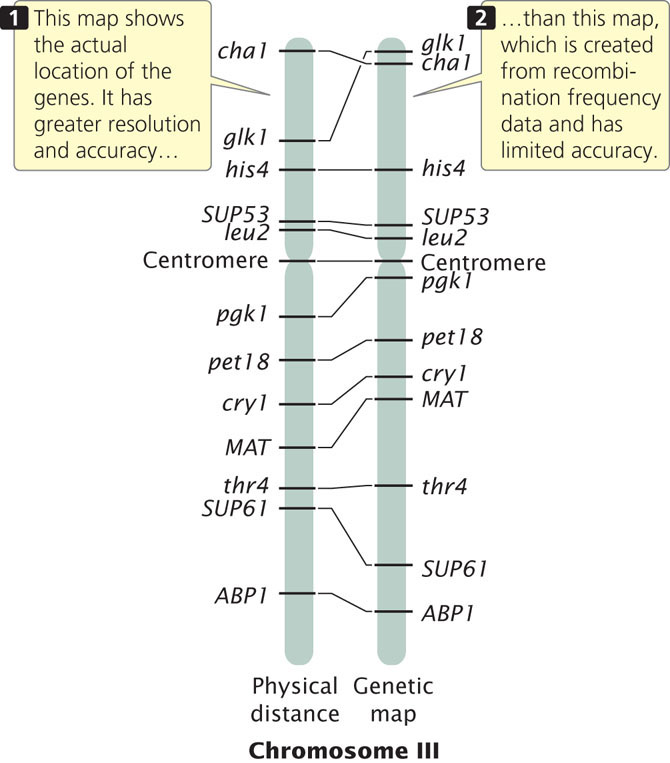
Genetic maps have several limitations, the first of which is resolution, or detail. The human genome includes 3.2 billion base pairs of DNA and has a total genetic distance of about 4000 cM, an average of 800,000 bp/cM. Even if a marker were present every centimorgan (which is unrealistic), the resolution in regard to the physical structure of the DNA would still be quite low. In other words, the detail of the map is very limited. A second problem with genetic maps is that they do not always accurately correspond to physical distances between genes. Genetic maps are based on rates of crossing over, which vary somewhat from one part of a chromosome to another; so the distances on a genetic map are only approximations of real physical distances along a chromosome. Figure 20.2 compares the genetic map of chromosome III of yeast with a physical map determined by DNA sequencing. There are some discrepancies between the distances and even among the positions of some genes. In spite of these limitations, genetic maps have been critical to the development of physical maps and the sequencing of whole genomes.
581
Physical Maps
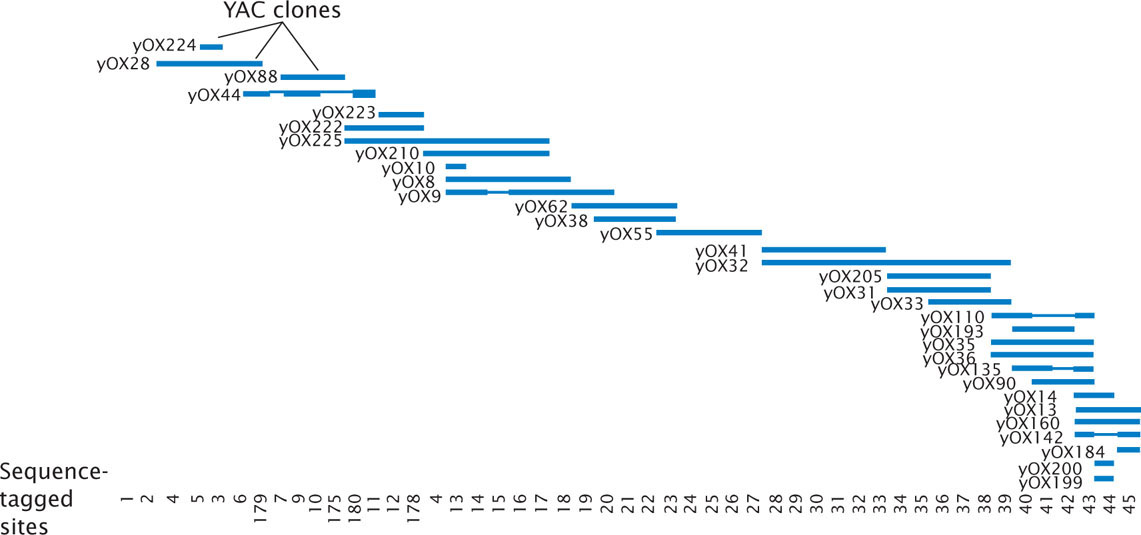
Physical maps are based on the direct analysis of DNA, and they place genes in relation to distances measured in number of base pairs, kilobases, or megabases. A common type of physical map is one that connects isolated pieces of genomic DNA that have been cloned in bacteria or yeast (Figure 20.3). Physical maps generally have higher resolution and are more accurate than genetic maps. A physical map is analogous to a neighborhood map that shows the location of every house along a street, whereas a genetic map is analogous to a highway map that shows the general locations of major towns and cities.
582
One of the techniques that has been used for creating physical maps is restriction mapping, which determines the position of restriction sites on DNA. When a piece of DNA is cut with a restriction enzyme and the fragments are separated by gel electrophoresis, the number of restriction sites in the DNA and the distances between them can be determined by the number and positions of bands on the gel. However, this information does not tell us the order or the precise location of the restriction sites. To map restriction sites, a sample of the DNA is cut with one restriction enzyme, and another sample is cut with a different restriction enzyme. A third sample is cut with both restriction enzymes together (a double digest). The DNA fragments produced by these restriction digests are then separated by gel electrophoresis, and their sizes are compared. Overlap in size of fragments produced by the digests can be used to position the restriction sites on the original DNA molecule (see the Worked Problem at the end of this chapter). Most restriction mapping is done with several restriction enzymes, used alone and in various combinations, producing many restriction fragments. With long pieces of DNA (greater than 30 kb), computer programs are used to determine the restriction maps, and restriction mapping may be facilitated by tagging one end of a large DNA fragment with radioactivity or by identifying the end with the use of a probe.
Physical maps, such as restriction maps of DNA fragments or even whole chromosomes, are often created for genomic analysis. These lengthy maps are created by combining maps of shorter, overlapping genomic fragments. A number of additional techniques exist for creating physical maps including: sequence-tagged site (STS) mapping, which locates the positions of short unique sequences of DNA on a chromosome; in situ hybridization, by which markers can be visually mapped to locations on chromosomes; and DNA sequencing.
CONCEPTS
Both genetic and physical maps provide information about the relative positions and distances between genes, molecular markers, and chromosome segments. Genetic maps are based on rates of recombination and are measured in percent recombination (map units) or centimorgans. Physical maps are based on physical distances and are measured in base pairs.
 CONCEPT CHECK 1
CONCEPT CHECK 1
What are some of the limitations of genetic maps?
Sequencing an Entire Genome
The first genomes to be sequenced were small virus genomes. The genome of bacteriophage λ, consisting of 49,000 bp, was completed in 1982. In 1995, the first genome of a free-living organism (Haemophilus influenzae) was sequenced by Craig Venter and Claire Fraser of The Institute for Genomic Research (TIGR) and Hamilton Smith of Johns Hopkins University. This bacterium has a small genome of 1.8 million base pairs (Figure 20.4). By 1996, the genome of the first eukaryotic organism (yeast) had been determined, followed by the genomes of Escherichia coli (1997), Caenorhabditis elegans (1998), Drosophila melanogaster (2000), and Arabidopsis thaliana (2000). The first draft of the human genome was completed in June 2000.
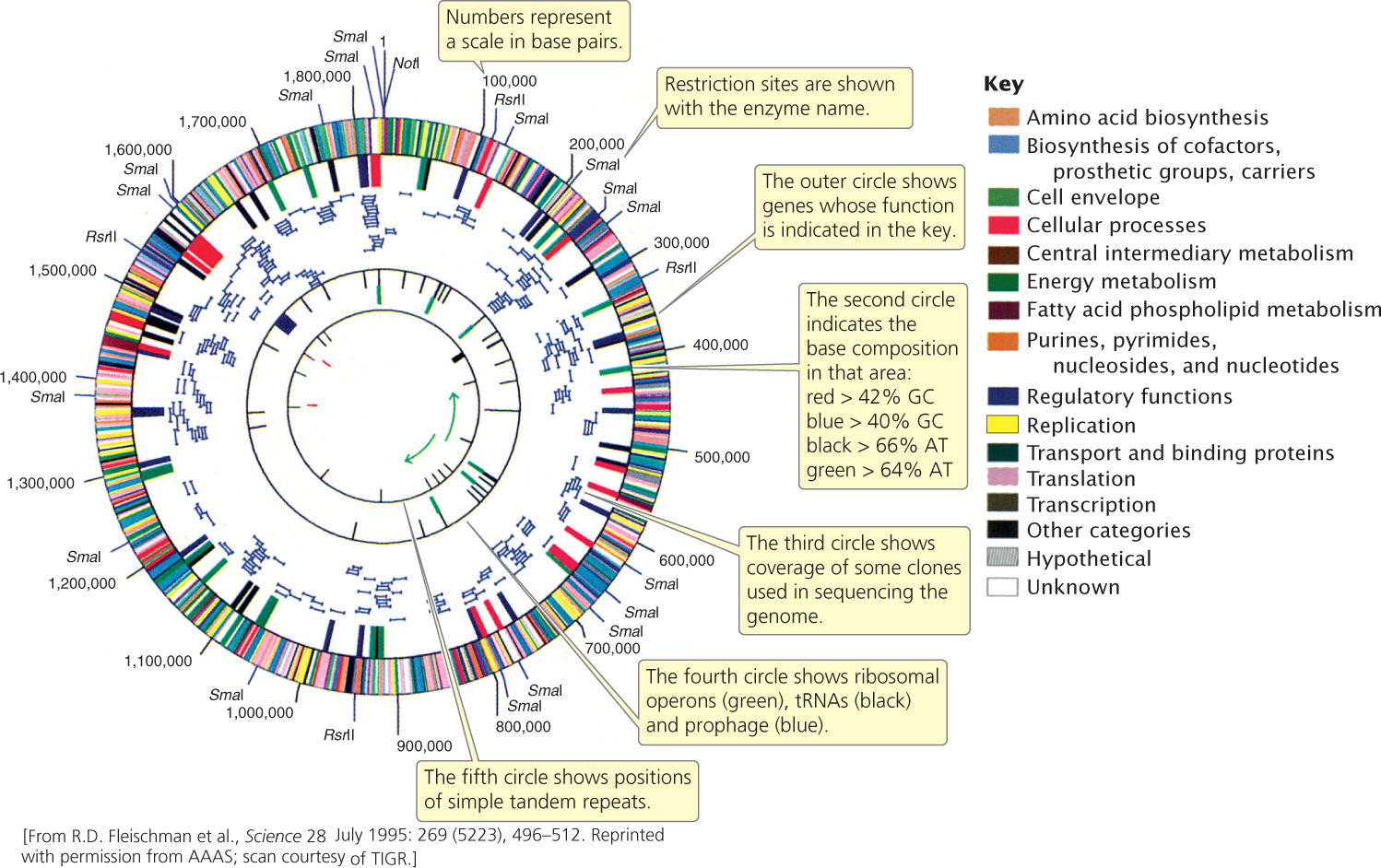
The ultimate goal of structural genomics is to determine the ordered nucleotide sequences of entire genomes of organisms. In Chapter 19, we considered some of the methods used to sequence small fragments of DNA. The main obstacle to sequencing a whole genome is the immense size of most genomes. Bacterial genomes are usually at least several million base pairs long; many eukaryotic genomes are billions of base pairs long and are distributed among dozens of chromosomes. Furthermore, for technical reasons, sequencing cannot begin at one end of a chromosome and continue straight through to the other end; only small fragments of DNA—usually no more than 500 to 700 nucleotides—can be sequenced at one time. Therefore, determining the sequence for an entire genome requires that the DNA be broken into thousands or millions of smaller fragments that can then be sequenced. The difficulty lies in putting these short sequences back together in the correct order. Two different approaches have been used to assemble the short sequenced fragments into a complete genome: map-based sequencing and whole-genome shotgun sequencing. We will consider these two approaches in the context of the Human Genome Project.
583
The Human Genome Project
By 1980, methods for mapping and sequencing DNA fragments had been sufficiently developed that geneticists began seriously proposing that the entire human genome could be sequenced. An international collaboration was planned to undertake the Human Genome Project (Figure 20.5); initial estimates suggested that 15 years and $3 billion would be required to accomplish the task.

The Human Genome Project officially began in October 1990. Initial efforts focused on developing new and automated methods for cloning and sequencing DNA and on generating detailed physical and genetic maps of the human genome. The effort was a public project consisting of the international collaboration of 20 research groups and hundreds of individual researchers who formed the International Human Genome Sequencing Consortium. This group used a map-based strategy for sequencing the human genome.
584
Map-Based Sequencing
In map-based sequencing, short sequenced fragments are assembled into a whole-genome sequence by first creating detailed genetic and physical maps of the genome, which provide known locations of genetic markers (restriction sites, other genes, or known DNA sequences) at regularly spaced intervals along each chromosome. These markers are later used to help align the short sequenced fragments into their correct order.
After the genetic and physical maps are available, chromosomes or large pieces of chromosomes are separated by pulsed-field gel electrophoresis (PFGE) or by flow cytometry. Standard gel electrophoresis cannot separate very large pieces of DNA, such as whole chromosomes, but PFGE can separate large molecules of DNA or whole chromosomes in a gel by periodically alternating the orientation of an electrical current. In flow cytometry, chromosomes are sorted optically by size.
Each chromosome (or sometimes the entire genome) is then cut up by partial digestion with restriction enzymes (Figure 20.6). Partial digestion means that the restriction enzymes are allowed to act for only a limited time so that not all restriction sites in every DNA molecule are cut. Thus, partial digestion produces a set of large overlapping DNA fragments, which are then cloned with the use of cosmids, yeast artificial chromosomes (YACs), or bacterial artificial chromosomes (BACs; see Chapter 19).
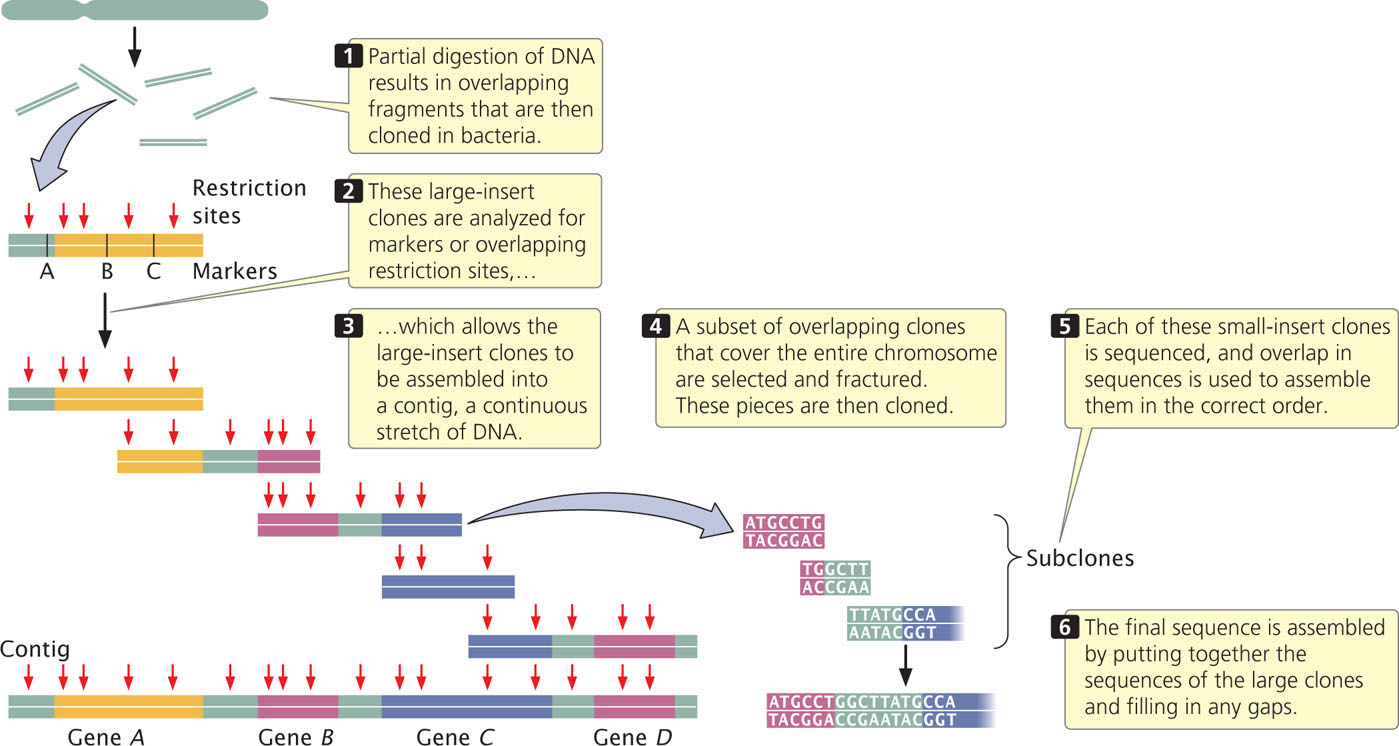
Next, these large-insert clones are put together in their correct order on the chromosome (see Figure 20.6). This assembly can be done in several ways. One method relies on the presence of a high-density map of genetic markers. A complementary DNA probe is made for each genetic marker, and a library of the large-insert clones is screened with the probe, which will hybridize to any colony containing a clone with the marker. The library is then screened for neighboring markers. Because the clones are much larger than the markers used as probes, some clones will have more than one marker. For example, clone A might have markers M1 and M2, clone B markers M2, M3, and M4, and clone C markers M4 and M5. Such a result would indicate that these clones contain areas of overlap, as shown here:
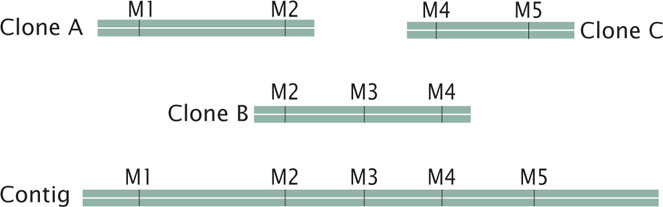
A set of two or more overlapping DNA fragments that form a contiguous stretch of DNA is called a contig. This approach was used in 1993 to create a contig consisting of 196 overlapping YAC clones (see Figure 20.3) of the human Y chromosome.
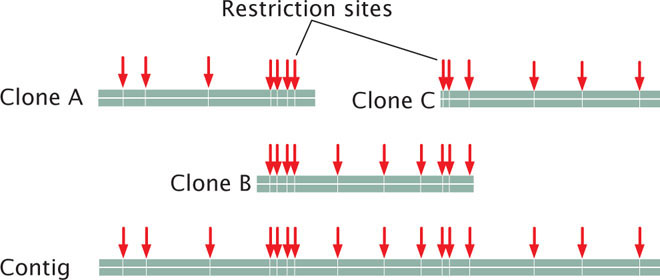
The order of clones can also be determined without the use of preexisting genetic maps. For example, each clone can be cut with a series of restriction enzymes and the resulting fragments then separated by gel electrophoresis. This method generates a unique set of restriction fragments, called a fingerprint, for each clone. The restriction patterns for the clones are stored in a database. A computer program is then used to examine the restriction patterns of all the clones and look for areas of overlap. The overlap is then used to arrange the clones in order, as shown here:
585
Other genetic markers can be used to help position contigs along the chromosome. ![]() TRY PROBLEM 27
TRY PROBLEM 27
When the large-insert clones have been assembled into the correct order on the chromosome, a subset of overlapping clones that efficiently cover the entire chromosome can be chosen for sequencing; the goal is to select the minimum number of clones that are necessary to represent the chromosome. Each of the selected large-insert clones is fractured into smaller overlapping fragments, which are themselves cloned (see Figure 20.6). These smaller clones (called small-insert clones) are then sequenced. The sequences of the small-insert clones are examined for overlap, which allows them to be correctly assembled to give the sequence of the large-insert clones. Enough overlapping small-insert clones are usually sequenced to ensure that the entire genome is sequenced several times. Finally, the whole genome is assembled by putting together the sequences of all overlapping contigs (see Figure 20.6). Often, gaps in the genome map still exist and must be filled in by using other methods.
The International Human Genome Sequencing Consortium used a similar map-based approach to sequencing the human genome. Many copies of the human genome were cut up into fragments of about 150,000 bp each, which were inserted into bacterial artificial chromosomes. Yeast artificial chromosomes and cosmids had been used in early stages of the project but did not prove to be as stable as the BAC clones, although YAC clones were instrumental in putting together some of the larger contigs. Restriction fingerprints were used to assemble the BAC clones into contigs, which were positioned on the chromosomes with the use of genetic markers and probes. The individual BAC clones were sheared into smaller overlapping fragments and sequenced, and the whole genome was assembled by putting together the sequence of the BAC clones.
Whole-Genome Shotgun Sequencing
In 1998, Craig Venter announced that he would lead a company called Celera Genomics in a private effort to sequence the human genome. He proposed using a shotgun sequencing approach, which he suggested would be quicker than the map-based approach employed by the International Human Genome Sequencing Consortium. In whole-genome shotgun sequencing (Figure 20.7), small-insert clones are prepared directly from genomic DNA and sequenced. Powerful computer programs then assemble the entire genome by examining overlap among the small-insert clones. One advantage of shotgun sequencing is that the small-insert clones can be placed into plasmids, which are simple and easy to manipulate. The requirement for overlap means that most of the genome will be sequenced multiple (often from 10 to 15) times. The average number of times a nucleotide in the genome is sequenced is called the sequencing coverage. For example, 10x coverage means an average nucleotide in the genome has been sequenced 10 times.
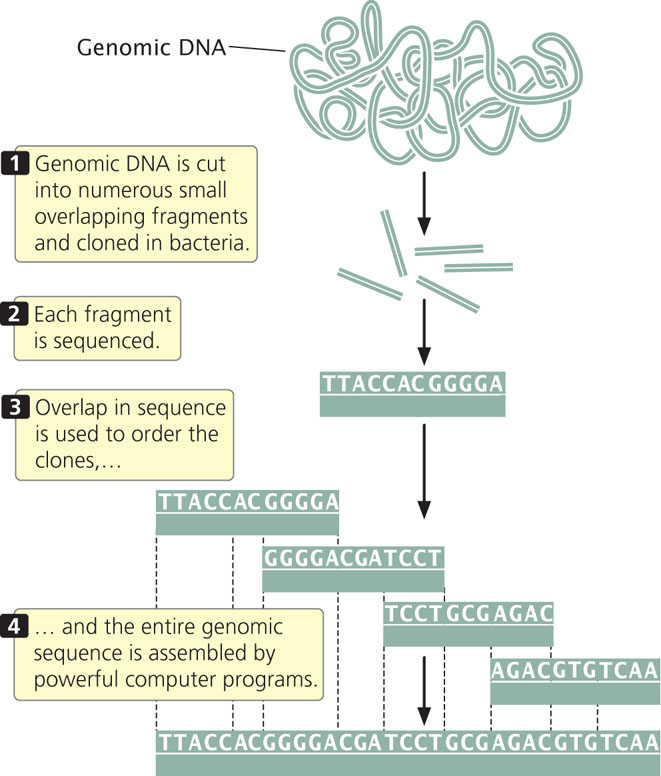
Shotgun sequencing was initially used for assembling small genomes such as those of bacteria. When Venter proposed the use of this approach for sequencing the human genome, it was not at all clear that the approach could successfully assemble a complex genome consisting of billions of base pairs such as the human genome. Today, virtually all genomes are sequenced using the whole-genome shotgun approach.
586
CONCEPTS
Sequencing a genome requires breaking it up into small overlapping fragments of which the DNA sequences can be determined in a sequencing reaction. In map-based sequencing, sequenced fragments are ordered into the final genome sequence with the use of genetic and physical maps. In whole-genome shotgun sequencing, the genome is assembled by comparing overlap in the sequences of small fragments.
CONCEPT CHECK 2
A contig is
- a set of molecular markers used in genetic mapping.
- a set of overlapping fragments that form a continuous stretch of DNA.
- a set of fragments generated by a restriction enzyme.
- a small DNA fragment used in sequencing.
Results and Implications of The Human Genome Project
In the summer of 2000, both public and private sequencing projects announced the completion of a rough draft that included most of the sequence of the human genome, 5 years ahead of schedule. The human genome sequence was declared completed in the spring of 2003, although some gaps still remain. For most chromosomes, the finished sequence is 99.999% accurate, with less than one base-pair error per 100,000 bp, an accuracy rate 10 times that of the initial goal.
With the first human genome determined, sequencing additional genomes is much easier. It is now possible to sequence an entire human genome in a single day, and genomes from thousands of different people have now been sequenced. The cost of sequencing a complete human genome has also dropped dramatically and will continue to fall as sequencing technology improves. A number of commercial companies are racing to develop the technology to sequence a complete human genome for $1,000 or less.
The availability of the complete sequence of the human genome is proving to be of great benefit. The sequence has provided tools for detecting and mapping genetic variants across the human genome, greatly facilitating gene mapping in humans. For example, several million sites at which people differ in a single nucleotide (called single-nucleotide polymorphisms; see next section) have now been identified, and these sites are being used in genome-wide association studies to locate genes that affect diseases and traits in humans. The sequence is also providing important information about development and many basic cellular processes.
Next-generation sequencing techniques that allow rapid and inexpensive sequencing of genomic DNA (see Chapter 19) are being used to address fundamental questions in many areas. For example, the genomes of a number of cancer cells have now been completely sequenced and compared with sequences of healthy cells from the same person, allowing complete determination of all the mutations that lead to tumor formation and cancer progression. The complete genome of an unborn baby was recently sequenced from fetal DNA isolated from its mother’s blood. The 1000 Genomes Project is sequencing and comparing the genomes of several thousand people from different ethnic groups, with the goal of detecting most of the common variations that exist in the human species. Sequencing of the complete genomes of parents and their children has allowed a direct estimate of mutation rates.
DNA has been extracted from the bones of ancient humans, including Neanderthals and Denisovans (a little-known group of humans that appear to be closely related to Neanderthals), and completely sequenced. Comparisons of the modern human genome with these and other species are adding to our understanding of human evolution, as well as our knowledge of the evolution and the history of all life.
In spite of these benefits and successes, some people have been disappointed by the lack of tangible results from the Human Genome Project. At the time it was proposed, there was speculation that the sequencing of the human genome would immediately revolutionize the practice of medicine, leading to new insights for treating common diseases and resulting in the development of powerful new drugs. Although genomic sequence data have produced numerous new and exciting research findings and has led to a better understanding of many diseases, the data are still seldom used by practicing physicians in the treatment of patients. Undoubtedly, genomic information will be important for medicine in the future, both for tailoring treatment to individual patients (personalized medicine) and for drug discovery, but when this will take place is presently uncertain.
Along with the many potential benefits of having complete sequence information, there are concerns about it being misused. With the knowledge gained from genomic sequencing, many more genes for diseases, disorders, and behavioral and physical traits will be identified, increasing the number of genetic tests that can be performed to make predictions about the future phenotype and health of a person. There is concern that information from genetic testing might be used to discriminate against people who are carriers of disease-causing genes or who might be at risk for some future disease. This matter has been addressed to some extent in the United States with the passage of the Genetic Information Nondiscrimination Act (see Chapter 6), which prohibits health insurers and employers from using genetic information to make decisions about health-insurance coverage and employment (although it does not apply to life, disability, and long-term care insurance). Questions also arise about who should have access to a person’s genome sequence. What about relatives, who have similar genomes and might also might be at risk for some of the same diseases? There are also questions about the use of this information to select for specific traits in future offspring. All of these concerns are legitimate and must be addressed if we are to use the information from genome sequencing responsibly.
587
CONCEPTS
The Human Genome Project was an effort to sequence the entire human genome. A rough draft of the sequence was completed by two competing teams, both of which finished a rough draft of the genome sequence in 2000. The entire sequence was completed in 2003. The ability to rapidly sequence human genomes raises a number of ethical questions.
Single-Nucleotide Polymorphisms
Since the completion of the sequencing of the human genome, sequencers have focused much of their effort on mapping differences among people in their genomic sequences.
Imagine that you are riding in an elevator with a random stranger. How much of your genome do you have in common with this person? Studies of variation in the human genome indicate that you and the stranger will be identical at about 99.9% of your DNA sequences. The difference between you and the stranger is very small in relative terms but, because the human genome is so large (3.2 billion base pairs), you and the stranger will be different at more than 3 million base pairs of your genomic DNA. These differences are what make each of us unique, and they greatly affect our physical features, our health, and possibly even our intelligence and personality.
A site in the genome where individual members of a species differ in a single base pair is called a single-nucleotide polymorphism (SNP, pronounced “snip”). Arising through mutation, SNPs are inherited as allelic variants (just as are alleles that produce phenotypic differences, such as blood types), although SNPs do not usually produce a phenotypic difference. Single-nucleotide polymorphisms are numerous and are present throughout genomes. In a comparison of the same chromosome from two different people, a SNP can be found approximately every 1000 bp.
Haplotypes and Linkage Disequilibrium
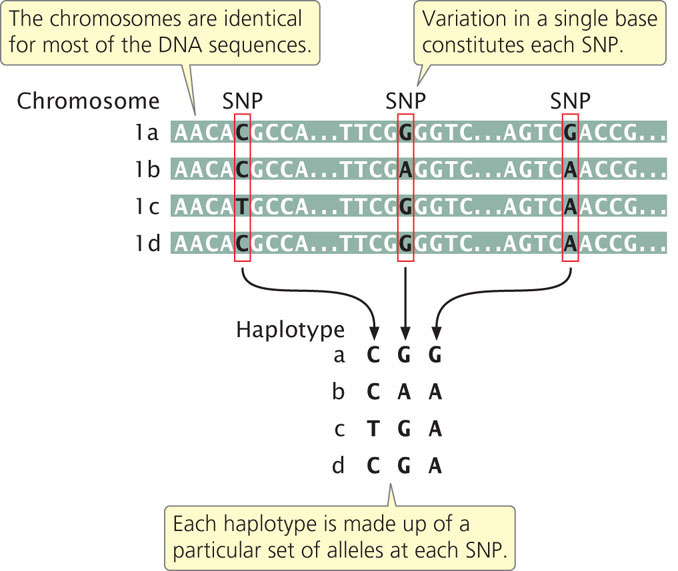
Most SNPs present within a population arose once from a single mutation that occurred on a particular chromosome and subsequently spread throughout the population. Thus, each SNP is initially associated with other SNPs (as well as other types of genetic variants or alleles) that were present on the particular chromosome on which the mutation arose. The specific set of SNPs and other genetic variants observed on a single chromosome or part of a chromosome is called a haplotype (Figure 20.8). SNPs within a haplotype are physically linked and therefore tend to be inherited together. New haplotypes can arise through mutation or crossing over, which breaks up the particular set of SNPs in a haplotype. Because the rate of crossing over is proportional to the physical distances between genes, SNPs and other genetic variants that are located close together on the chromosome will be strongly associated as haplotypes. The nonrandom association between genetic variants within a haplotype is called linkage disequilibrium. Because the SNPs in a haplotype are inherited together, a haplotype consisting of thousands of SNPs can be identified with the use of only a few SNPs. The few SNPs used to identify a haplotype are called tag SNPs. There are about 10 million SNPs in the human population but, because of linkage disequilibrium, these SNPs are present in a much smaller number of haplotypes. Therefore, a relatively small number of SNPs—perhaps only 100,000— can be used to identify most of the haplotypes in humans.
The Use of Single-Nucleotide Polymorphisms
Because of their variability and widespread occurrence throughout the genome, SNPs are valuable as markers in linkage studies. When a SNP is physically close to a disease-causing locus, it will tend to be inherited along with the disease-causing allele. People with the disease will tend to have different SNPs from those of healthy people. A comparison of SNP haplotypes in people with a disease and in healthy people can reveal the presence of genes that affect the disease; because the disease gene and the SNP are closely linked, the location of the disease-causing gene can be determined from the location of associated SNPs. This approach is the same as that used in gene mapping with RFLPs (see Chapter 19), but there are many more SNPs than RFLPs, providing a dense set of variable markers covering the entire genome that can be used more effectively in mapping.
An international effort, called the International HapMap Project, was begun in 2002 with the goal of cataloging and mapping SNPs and other genetic variants that could be used to identify common haplotypes in human populations for use in linkage and family studies. Phase I of the project, completed in 2005, cataloged more than 1 million SNPs in the genomes of 269 people from four diverse human populations (African, Japanese, Chinese, and European). These SNPs are spread over all 23 human chromosomes at a distance of about one SNP per 5000 bp. Phase II of the project, completed in 2006, cataloged a total of 4.6 million SNPs.
588
Alleles from most of the common SNPs are found in all four human ethnic groups, although the frequencies of the alleles vary considerably among human populations. The greatest genetic diversity of SNPs is found within Africans, which is consistent with many other studies that suggest that humans first evolved in Africa.
Data from the HapMap Project have provided important information about the function and evolution of the human genome. For example, studies of linkage disequilibrium with the use of SNPs have determined that recombination does not take place randomly across the chromosomes: there are numerous recombination hotspots, where more recombination takes place than expected merely by chance. The distribution of SNPs is also providing information about regions of the human genome that have evolved quickly in the recent past, providing clues to how humans have responded to natural selection.
Genome-Wide Association Studies
Many common diseases are caused by complex interactions among multiple genes: the availability of SNPs has greatly facilitated the search for these genes. Genome-wide association studies use numerous SNPs scattered across the genome to find genes of interest. Soon after the completion of phase I of the HapMap Project, researchers used the SNP data to conduct a genome-wide association study for the presence of a major gene that causes age-related macular degeneration, one of the leading causes of blindness among the elderly. In this study, researchers genotyped 96 people with macular degeneration and 50 people with healthy eyes for more than 100,000 SNPs that blanketed the genome. The study revealed a strong association between the disease and a gene on chromosome 1 that encodes complement factor H, which has a role in immune function. This finding suggests that macular degeneration might be treated with drugs that affect complement proteins. Another study uncovered a major gene associated with Crohn disease, a common inflammatory disorder of the gastrointestinal tract. Other studies using genome-wide scans of SNPs have identified genes that contribute to heart disease, bone density, prostate cancer, and diabetes.
In one successful application of SNPs for finding disease associations, researchers genotyped 17,000 people in the United Kingdom for 500,000 SNPs in 2007. They detected strong associations between 24 genes and chromosome segments and the incidence of seven common diseases, including coronary artery disease, Crohn disease, rheumatoid arthritis, bipolar disorder, hypertension, and two types of diabetes. The importance of this study is its demonstration that genome-wide association studies utilizing SNPs could successfully locate genes that contribute to complex diseases caused by multiple genetic and environmental factors.
Within the past few years, SNPs have been used in genome-wide association studies to successfully locate genes that influence many additional traits, such as the age of puberty and menopause in women, variation in facial features, skin pigmentation, eye color, body weight, bone density, glaucoma, and even susceptibility to infectious diseases such as meningococcal disease and tuberculosis. Unfortunately, the genes identified often explain only a small proportion of the genetic influence on the trait. For example, one huge genome-wide association study combined data from over 100,000 human subjects in an attempt to locate genes coding for blood lipids and cardiovascular disease. Although the study identified 95 different loci associated with lipid traits, these genes only correspond to 25-30% of the total genetic variation in these traits.
DNA sequences that encode the majority of the missing genetic variation in such traits—sometimes called the “dark matter of the genome”—have, this far, remained largely undetected. The low percentage of variation explained by most current genome-wide association studies means that the genes identified are not, by themselves, useful predictors of the risk of inheriting the disease or trait. Nevertheless the identification of specific genes that influence a disease or trait can lead to a better understanding of the biological processes that produce the phenotype.
Copy-Number Variations
A diploid person normally possesses two copies of every gene, one inherited from the mother and one inherited from the father. Nevertheless, studies of the human genome have revealed differences among people in the number of copies of large DNA sequences (greater than 1000 bp); these variations are called copy-number variations (CNVs). Copy-number variations may include deletions, causing some people to have only a single copy of a sequence, or duplications, causing some people to have more than two copies. A study of CNVs in 270 people from four populations (those studied in the HapMap Project) identified a surprising number of these types of variants: more than 1447 genomic regions varied in copy number, encompassing 12% of the human genome. But only a small part of the human genome was surveyed in this study, and so the CNVs detected are only a small subset of all that exist. Many of the CNVs encompassed large regions of DNA sequence, often several hundred thousand base pairs in length. Thus, people differ not only at millions of individual SNPs, but also in the number of copies of many larger segments of the genome.
Most CNVs contain multiple genes and potentially affect the phenotype by altering gene dosage and by changing the position of sequences, which may affect the regulation of nearby genes. Indeed, several studies have found associations between CNVs and disease, and even between CNVs and normal phenotypic variability in human populations. For example, variations in the number of copies of the UGT2B17 gene contribute to differences in testosterone metabolism among individuals and affect the risk of prostate cancer. Copy-number variations have been associated with Crohn disease, rheumatoid arthritis, psoriasis, schizophrenia, autism, diabetes, and intellectual disability.
589
Sequence-Tagged Sites and Expressed-Sequence Tags
A sequence-tagged site (STS) is a short (200-500 bp) sequence of DNA, present only once in the genome, whose chromosomal location has been determined. STSs are often used to determine the genomic location of a DNA clone. Researchers have developed PCR primers for each STS, allowing the amplification of the STS by PCR. In this way, PCR can be used to detect the presence of an STS in a set of clones. Because the chromosomal location of the STS is known, the presence of the STS in a particular clone provides information about where in the genome that clone originated.
Another type of identified sequence is an expressed-sequence tag (EST). These are sequences corresponding to DNA that is transcribed into mRNA. In most eukaryotic organisms, only a small percentage of the DNA encodes proteins; for example, in humans, about 1.5% of the DNA encodes proteins. If only protein-encoding genes are of interest, an examination of mRNA rather than the entire DNA genomic sequence is often more efficient. Expressed-sequence tags are obtained by isolating RNA from a cell and subjecting it to reverse transcription, producing a set of cDNA fragments that correspond to RNA molecules from the cell. Short stretches from the ends of these cDNA fragments are then sequenced, and the sequence obtained (called a tag) provides a marker that identifies the DNA fragment. Expressed-sequence tags can be used to find active genes in a particular tissue or at a particular point in development.
CONCEPTS
In addition to collecting genomic-sequence data, genomic projects are collecting databases of nucleotides that vary among individual organisms (single-nucleotide polymorphisms, SNPs), variations in the number of copies of sequences (copy-number variations), unique sequences whose chromosomal location has been mapped (sequence-tagged sites, STSs), and markers associated with transcribed sequences (expressed-sequence tags, ESTs).
CONCEPT CHECK 3
What was the goal of the HapMap Project?
Bioinformatics
Complete genome sequences have now been determined for numerous organisms, with many additional projects underway. These studies are producing tremendous quantities of sequence data. Cataloging, storing, retrieving, and analyzing this huge data set are major challenges of modern genetics. Bioinformatics is an emerging field consisting of molecular biology and computer science that centers on the development of databases, computer-search algorithms, gene-prediction software, and other analytical tools that are used to make sense of DNA-, RNA-, and protein-sequence data. Bioinformatics develops and applies these tools to “mine the data,” extracting the useful information from sequencing projects. The development and use of algorithms and computer software for analyzing DNA- and protein-sequence data have helped to make molecular biology a more-quantitative field. Sequence data in publicly available online databases enable scientists and students throughout the world to access this tremendous resource.
A number of databases have been established for the collection and analysis of DNA- and protein-sequence information. Primary databases contain the sequence information, along with information that describes the source of the sequence and its determination. Secondary databases contain the results of analyses carried out on the primary sequence data, such as information about particular sequence patterns, variations, mutations, and evolutionary relationships.
After a genome has been sequenced, one of the first tasks is to identify potential genes within the sequence. Unfortunately, there are no universal characteristics that mark the beginning and end of a gene. The enormous amount of DNA in a genome and the complexities of gene structure make finding genes within the sequence a difficult task. Computer programs have been developed to look for specific sequences in DNA that are associated with certain genes. There are two general approaches to finding genes. The ab initio approach scans the sequence looking for features that are usually within a gene. For example, protein-encoding genes are characterized by an open reading frame, which includes a start codon and a stop codon in the same reading frame. Specific sequences mark the splice sites at the beginning and end of introns; other specific sequences are present in promoters immediately upstream of start codons. The comparative approach looks for similarity between a new sequence and sequences of all known genes. If a match is found, then the new sequence is assumed to be a similar gene. Some of these computer programs are capable of examining databases of EST and protein sequences to see if there is evidence that a potential gene is expressed.
It is important to note that the programs that have been developed to identify genes on the basis of DNA sequence are not perfect. Therefore, the numbers of genes reported in most genome projects are estimates. The presence of multiple introns, alternative splicing, multiple copies of some genes, and much noncoding DNA between genes makes accurate identification and counting of genes difficult.
After a gene has been identified, it must be annotated, which means linking its sequence information to other information about its function and expression, the protein that it encodes, and information on similar genes in other species. There are a number of methods of probing a gene’s function, which will be discussed in the Section 20.2 on functional genomics. Computer programs are available for determining whether similar sequences have already been found, either in the same species or in different species. The most widely used of these programs is the Basic Local Alignment Search Tool (BLAST). To conduct a BLAST search, a researcher submits a query sequence and the program searches the database for any other sequences that have regions of high similarity to the query sequence. The program returns all sequences in the database that are similar, along with information about the degree of similarity and the significance of the match (how likely the similarity would be to occur by chance alone).
590
Additional databases contain information on sequence diversity—how and where a genome varies among individual organisms. The human genome has now been mapped for millions of SNPs, and information about the frequency of these variants in different populations is being collected. Additional databases have been developed to catalog all known mutations causing particular diseases; the Human Variome Project is an effort to collect and make available all genetic variations that affect human health. Important information about the expression patterns of the thousands of genes found in genomes also is being compiled. ![]() TRY PROBLEM 29
TRY PROBLEM 29
CONCEPTS
Bioinformatics is an interdisciplinary field that combines molecular biology and computer science. It develops databases of DNA and protein sequences and tools for analyzing those sequences.
CONCEPT CHECK 4
The ab initio approach finds genes by looking for
- common sequences found in most genes.
- similarity in sequence with known genes.
- mRNA with the use of in situ hybridization.
- mutant phenotypes.
Metagenomics
Advances in sequencing technology, which have made sequencing faster and less expensive, now provide the possibility of sequencing not just the genomes of individual species but the genomes of entire communities of organisms. Metagenomics is an emerging field in which the genome sequences of an entire group of organisms that inhabit a common environment are sampled and determined.
Thus far, metagenomics has been applied largely to microbial communities. Traditionally, bacteria have been studied by growing and analyzing them in the laboratory. However, many bacteria cannot be cultured with the use of laboratory techniques. Metagenomics analyzes microbial communities by extracting DNA from the environment, determining its sequences, and reconstructing community composition and function on the basis of genome sequences. This technique allows the identification and genetic analysis of species that cannot be grown in the laboratory and have never been studied by traditional microbiological methods. The entire genomes of some dominant species have been reconstructed from environmental samples, providing scientists with a great deal of information on the biology of these microbes.
An early metagenomic study analyzed the microbial community found in acid drainage from a mine and determined that this community consisted of only a few dominant bacterial species. Another study, called the Global Ocean Sampling Expedition, followed the route of Darwin’s voyage on the H.M.S. Beagle in the 1800s. Scientists collected ocean samples and used metagenomic methods to determine their microbial communities. In this study, scientists cataloged sequences for more than 6 million proteins, including more than 1700 new protein families. Some important results have already emerged from metagenomic studies. Analyses of bacterial genomes found in ocean samples led to the discovery of proteorhodopsin proteins, which are light-driven proton pumps. Subsequent research demonstrated that these proteins are found in diverse microbial groups and are a major source of energy flux in the world’s oceans.
Other metagenomic studies have examined the genes of bacteria that inhabit the human intestinal tract. These bacteria, along with those that inhabit the skin and other parts of the human body, are termed the human microbiome. The microbiome of a typical person includes over 100 trillion cells—more than 10 times as many as human cells—and contains 100 times as many genes as the human genome.
Research is demonstrating that the human microbiome plays an important role in human health. One study examined the gut microflora of obese and lean people. Two groups of bacteria are common in the human gut: Bacteroidetes and Firmicutes. Researchers discovered that obese people have relatively more Firmicutes than do lean people and that the proportion of Firmicutes decreases in obese people who lose weight on a low-calorie diet. These same results were observed in obese and lean mice. In an elegant experiment, researchers transferred bacteria from obese to lean mice. The mice that received bacteria from obese mice extracted more calories from their food and stored more fat, suggesting that gut microflora might play some role in obesity.
Ecological communities have traditionally been characterized on the basis of the species that they contain. Metagenomics affords the possibility of characterizing communities on the basis of their component genes, an approach that has been termed gene-centric. A gene-centric approach leads to new questions: Are certain types of genes more common in some communities than others? Are some genes essential for energy flow and nutrient recycling within a community? Because of the larger sizes of their genomes, eukaryotic communities have not yet been the focus of these approaches, but many researchers predict that they will be in the future.
591
CONCEPTS
Metagenomic studies examine the genomes of communities of organisms that inhabit a common environment. This approach has been applied to microbial communities and allows the composition and genetic makeup of the community to be determined without cultivating and isolating individual species. Metagenomic studies are a source of important new insights into microbial communities.
Synthetic Biology
The ability to sequence and study whole genomes, coupled with an increased understanding of the genetic information required for basic biological processes, now provides the possibility of creating—entirely from scratch—novel organisms that have never before existed. Synthetic biology is a new field that seeks to design organisms that might provide useful functions, such as microbes that provide clean energy or break down toxic wastes.
Synthetic biologists have already mixed and matched parts from different organisms to synthesize microbes. In 2002, geneticists recreated the poliovirus by joining together pieces of DNA that were synthesized in the laboratory. Even more impressively, in 2010 Daniel Gibson and his colleagues synthesized from scratch the complete 1.08-million-base-pair genome of the bacterium Mycoplasma mycoides. They started with a thousand pieces of DNA that were synthesized in the laboratory and then joined them together in successively larger pieces until they had assembled a complete copy of the genome. Within their synthetic genome they included a set of DNA sequences that spelled out—in code—an email address, the names of the researchers who participated in the project, and several well-known quotations. Finally, the researchers transplanted the artificial genome into a cell of a different bacterial species, M. capricolum, whose original genome had been removed. The new cell then began expressing the traits specified by the synthetic genome.
Synthetic biology has also been extended to eukaryotic cells. In 2011, geneticists replaced 90,000 bp of DNA of yeast chromosome 9 and 30,000 bp of yeast chromosome 6 with synthetic DNA. These partially synthetic yeast cells grew normally and exhibited only minor differences in gene expression. The ultimate goal of these experiments is to replace the entire 12 million bp genome of yeast with human-designed sequences.
These types of experiments have raised a number of concerns. The ability to tailor-make novel genomes and mix and match parts from different organisms creates the potential for the synthesis of dangerous microbes that might create ecological havoc if they escape from the laboratory or that could be used in biological warfare or bioterrorism. Ongoing discussions among geneticists, ethicists, security experts, and politicians are addressing these concerns and whether synthetic genomes can be safely made and used.