20.3 Comparative Genomics Studies How Genomes Evolve
Genome-sequencing projects provide detailed information about gene content and organization in different species and even in different members of the same species, allowing inferences about how genes function and genomes evolve. They also provide important information about evolutionary relationships among organisms and about factors that influence the speed and direction of evolution. Comparative genomics is the field of genomics that compares similarities and differences in gene content, function, and organization among genomes of different organisms.
Prokaryotic Genomes
More than 2000 prokaryotic genomes have now been sequenced. Most prokaryotic genomes consist of a single circular chromosome. However, there are exceptions, such as Vibrio cholerae, the bacterium that causes cholera, which has two circular chromosomes, and Borrelia burgdorferi, which has one large linear chromosome and 21 smaller chromosomes.
Genome Size and Number of Genes
The total amount of DNA in prokaryotic genomes ranges from 490,885 bp in Nanoarchaeum equitans, an archaea that lives entirely within another archaea, to 9,105,828 bp in Bradyrhizobium japonicum, a soil bacterium (Table 20.1). Although this range in genome size might seem extensive, it is much less than the enormous range of genome sizes seen in eukaryotes, which can vary from a few million base pairs to hundreds of billions of base pairs. Escherichia coli, the most widely used bacterium for genetic studies, has a fairly typical genome size at 4.6 million base pairs. Archaea and bacteria are similar in their ranges of genome size. Surprisingly, genome size also varies extensively within some species; for example, different strains of E. coli vary in genome size by more than 1 million base pairs.
597
| Species | Size (millions of base pairs) | Number of Predicted Genes |
|---|---|---|
| Archaea | ||
| Archaeoglobus fulgidus | 2.18 | 2407 |
| Methanobacterium thermoautotrophicum | 1.75 | 1869 |
| Nanoarchaeum equitans | 0.490 | 536 |
| Eubacteria | ||
| Bacillus subtilis | 4.21 | 4100 |
| Bradyrhizobium japonicum | 9.11 | 8317 |
| Escherichia coli | 4.64 | 4289 |
| Haemophilus influenzae | 1.83 | 1709 |
| Mycobacterium tuberculosis | 4.41 | 3918 |
| Mycoplasma genitalium | 0.58 | 480 |
| Staphylococcus aureus | 2.88 | 2697 |
| Vibrio cholerae | 4.03 | 3828 |
Source: Data from the Genome Atlas of the Center for Biological Sequence Analysis, www.cbs.dtu.dk/services/GenomeAtlas.
Among prokaryotes, the number of genes typically varies from 1000 to 2000, but some species have as many as 6700 and others as few as 480. Interestingly, the density of genes is rather constant across all species, with an average of about one gene per 1000 bp. Thus, prokaryotes with larger genomes will have more genes, in contrast with eukaryotes, for which there is little association between genome size and number of genes (see the next section). The evolutionary factors that determine the size of prokaryotic genomes (as well as eukaryotic genomes) are still largely unknown. However, the time required for cell division is frequently longer in organisms with larger genomes because of the time required to replicate the DNA before division. Thus, selection may favor smaller genomes in organisms that occupy environments where rapid reproduction is advantageous.
Prokaryotes with the smallest genomes tend to be in species that occupy restricted habitats, such as bacteria that live inside other organisms. The constant environment and metabolic functions supplied by the host organism may allow these bacteria to survive with fewer genes. Bacteria with the larger genomes tend to occupy highly complex and variable environments, such as the soil or the root nodules of plants. In these environments, genes that are needed only occasionally may be useful. In complex environments where resources are abundant, there may be little need for rapid division (which favors smaller genomes).
An example of a large-genome bacterium occupying a complex environment is Streptomyces coeliocolor, a filamentous bacterium with a genome size of 8,677,507 bp. This bacterium has been called the “Boy Scout” bacterium because it has a diverse set of genes and therefore follows the Scout motto: be prepared. In addition to the usual housekeeping genes for genetic functions such as replication, transcription, and translation, its genome contains a number of genes for the breakdown of complex carbohydrates, allowing it to consume decaying matter from plants, animals, insects, fungi, and other bacteria. It has genes for a large number of proteins that produce secondary metabolites (breakdown products) that can function as antibiotics and protect against desiccation and low temperatures. In this species, a large genome with lots of genes appears to be beneficial in the complex environment that it inhabits.
Horizontal Gene Transfer
Bacteria possess a number of mechanisms by which they can gain and lose DNA. DNA can be lost through simple deletion and can be gained by gene duplication and through the insertion of transposable genetic elements. Another mechanism for gaining new genetic information is horizontal gene transfer, a process by which both closely and distantly related bacterial species periodically exchange genetic information over evolutionary time. Such exchange may take place through the bacterial uptake of DNA in the environment (transformation), through the exchange of plasmids, and through viral vectors (see Chapter 9). Horizontal gene transfer has been recognized for some time, but analyses of many microbial genomes now indicate that it is more extensive than was formerly recognized. The widespread occurrence of horizontal transfer has caused some biologists to question whether distinct species even exist in bacteria (see Chapter 9 for more discussion of this matter).
598
Function of Genes
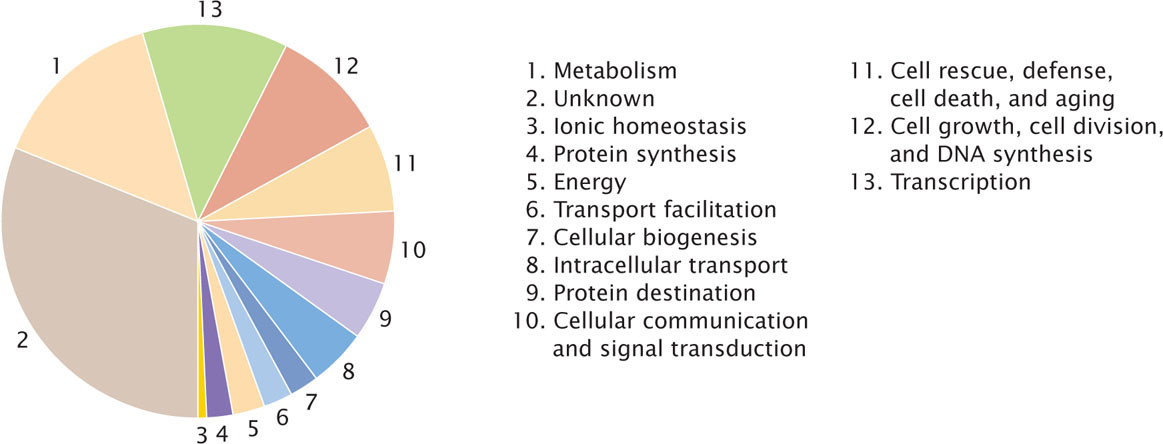
Only about half of the genes identified in prokaryotic genomes can be assigned a function. Almost a quarter of the genes have no significant sequence similarity to any known genes in other bacteria. The number of genes that encode biological functions such as transcription and translation tends to be similar among species, even when their genomes differ greatly in size. This similarity suggests that these functions are encoded by a basic set of genes that does not vary among species. In contrast, the number of genes taking part in biosynthesis, energy metabolism, transport, and regulatory functions varies greatly among species and tends to be higher in species with larger genomes. The functions of predicted genes (i.e., genes identified by computer programs) and known genes in E. coli are presented in Figure 20.14. A substantial part of the “extra” DNA found in the larger bacterial genomes is made up of paralogous genes that have arisen by duplication.
CONCEPTS
Comparative genomics compares the content and organization of whole genomic sequences from different organisms. Prokaryotic genomes are small, usually ranging from 1 million to 3 million base pairs of DNA, with several thousand genes. Prokaryotes with the smallest genomes tend to occupy restricted habitats, whereas those with the largest genomes are usually found in complex environments. Horizontal gene transfer has played a major role in bacterial genome evolution.
 CONCEPT CHECK 7
CONCEPT CHECK 7
What is the relation between genome size and gene number in prokaryotes?
Eukaryotic Genomes
The genomes of more than 150 eukaryotic organisms have been completely sequenced, including a number of fungi and protists, several insects, almost 30 species of plants, and numerous vertebrates. Sequenced eukaryotes include papayas, corn, rice, sorghum, grapevine, silkworms, several fruit flies, aphids, mosquitoes, anemones, mice, rats, dogs, cows, horses, orangutans, chimpanzees, and humans. Even the genomes of some extinct organisms have now been sequenced, including those of the wooly mammoth and Neanderthals. Hundreds of additional eukaryotic genomes are in the process of being sequenced. It is important to note that, even though the genomes of these organisms have been “completely sequenced,” many of the final assembled sequences contain gaps, and regions of heterochromatin may not have been sequenced at all. Thus, the sizes of eukaryotic genomes are often estimates, and the number of base pairs given for the genome size of a particular species may vary. Predicting the number of genes that are present in a genome also is difficult and may vary, depending on the assumptions made and the particular gene-finding software used.
Genome Size and Number of Genes
The genomes of eukaryotic organisms (Table 20.2) are larger than those of prokaryotes, and, in general, multicellular eukaryotes have more DNA than do simple, single-celled eukaryotes such as yeast. However, there is no close relation between genome size and complexity among the multicellular eukaryotes. For example, the mosquito (Anopheles gambiae) and fruit fly (Drosophila melanogaster) are both insects with similar structural complexity, yet the mosquito has 60% more DNA than the fruit fly.
| Species | Genome Size (millions of base pairs) | Number of Predicted Genes |
|---|---|---|
| Saccharomyces cerevisiae (yeast) | 12 | 6,144 |
| Physcomitrella patens (moss) | 480 | 38,354 |
| Arabidopsis thaliana (mustard plant) | 125 | 25,706 |
| Zea mays (corn) | 2,400 | 32,000 |
| Hordeum vulgare (barley) | 5,100 | 26,159 |
| Caenorhabditis elegans (roundworm) | 103 | 20,598 |
| Drosophila melanogaster (fruit fly) | 170 | 13,525 |
| Anopheles gambiae (mosquito) | 278 | 14,707 |
| Danio rerio (zebraflsh) | 1,465 | 22,409 |
| Takifugu rubripes (tiger pufferflsh) | 329 | 22,089 |
| Xenopus tropicalis (clawed frog) | 1,510 | 18,429 |
| Anolis carolinesis (anole lizard) | 1,780 | 17,792 |
| Mus musculus (mouse) | 2,627 | 26,762 |
| Pan troglodytes (chimpanzee) | 2,733 | 22,524 |
| Homo sapiens (human) | 3,223 | ~20,000 |
Source: Ensembl Web site: http://useast.ensembl.org/index.html and plants.ensembl.org/index.html
In general, eukaryotic genomes also contain more genes than do prokaryotes (but some large bacteria have more genes than single-celled yeasts have), and the genomes of multicellular eukaryotes have more genes than do the genomes of single-celled eukaryotes. In contrast with bacteria, there is no correlation between genome size and number of genes in eukaryotes. The number of genes among multicellular eukaryotes also is not obviously related to phenotypic complexity: humans have more genes than do invertebrates but only twice as many as fruit flies and fewer than the plant A. thaliana. The nematode C. elegans has more genes than D. melanogaster but is less complex. Additionally, the pufferfish has only about one-tenth the amount of DNA present in humans and mice but about as many genes. Eukaryotic genomes contain multiple copies of many genes, indicating that gene duplication has been an important process in genome evolution.
599
Most of the DNA in multicellular organisms is noncoding, and many genes are interrupted by introns. In the more complex eukaryotes, both the number and the length of the introns are greater.
Segmental Duplications and Multigene Families
Many eukaryotic genomes, especially those of multicellular organisms, are filled with segmental duplications, regions greater than 1000 bp that are almost identical in sequence. For example, about 4% of the human genome consists of segmental duplications. In most segmental duplications, the two copies are found on the same chromosome (an intrachromosomal duplication), but, in others, the two copies are found on different chromosomes (an interchro-mosomal duplication). In the human genome, the average size of segmental duplications is 15,000 bp.
Segmental duplications arise from processes that generate chromosome duplications, such as unequal crossing over (see Chapter 8). After a segmental duplication arises, it promotes further duplication by the occurrence of misalignment among the duplicated regions. Segmental duplication plays an important role in evolution by giving rise to new genes. After a segmental duplication arises, the original copy of the gene can continue its function while the new copy undergoes mutation. These changes may eventually lead to new function. The importance of gene duplication in genome evolution is demonstrated by the large number of multigene families that exist in many eukaryotic genomes. A multigene family is a group of evolutionarily related genes that arose through repeated evolution of an ancestral gene. For example, the globin gene family in humans consists of 13 genes that encode globinlike molecules, most of which produce proteins that carry oxygen. An even more spectacular example is the human olfactory multigene family, which consists of about 1000 genes that encode olfactory receptor molecules used in our sense of smell.
Noncoding DNA
Most eukaryotic organisms contain vast amounts of DNA that do not encode proteins. For example, only about 1.5 percent of the human genome consists of DNA that directly specifies the amino acids of proteins. The function of the remainder of DNA sequences, called noncoding DNA, has long been in question. Some research has suggested that much of this DNA has no function. For example, Marcelo Nobrega and his colleagues genetically engineered mice that were missing a large chromosomal region with no protein-encoding genes (called a gene desert). In one experiment, they created mice that were missing a 1,500,000-bp gene desert from mouse chromosome 3; in another, they created mice missing an 845,000-bp gene desert from chromosome 19. Remarkably, these mice appeared healthy and were indistinguishable from control mice. The researchers concluded that large regions of the mammalian genome can be deleted without major phenotypic effects and may, in fact, be superfluous.
600
However, other research suggested that gene deserts may contain sequences that have a functional role. For example, genome-wide association studies demonstrated that DNA sequences contained within a gene desert on human chromosome 9 are associated with coronary artery disease, and subsequent studies have demonstrated the presence of 33 enhancers in this gene desert.
In 2002 the Encyclopedia of DNA Elements (ENCODE) project was undertaken to determine if noncoding DNA had any function. Researchers cataloged all nucleotides within the genome that provide some function, including sequences encoding proteins and RNA molecules, and those serving as control sites for gene expression. This 10-year project was carried out by a team of over 400 scientists from around the world. In a series of papers published in 2012, ENCODE concluded that at least 80% of the human genome is involved in some type of gene function. Many of the functional sequences consisted of sites where proteins bind and influence the expression of genes. Prior to this study, much of the genome was considered “junk DNA” with no function, but the ENCODE study has greatly altered this view and suggests that there is little nonfunctional DNA in the human genome.
Transposable Elements
A substantial part of the genomes of most multicellular organisms consists of moderately and highly repetitive sequences (see Chapter 11), and the percentage of repetitive sequences is usually higher in those species with larger genomes (Table 20.3). Most of these repetitive sequences appear to have arisen through transposition. In the human genome, 45% of the DNA is derived from transposable elements, many of which are defective and no longer able to move. In corn, 85% of the genome is derived from transposable elements.
| Organism | Percentage of Genome |
|---|---|
| Arabidopsis thaliana (plant) | 10.5 |
| Zea mays (corn) | 85.0 |
| Caenorhabditis elegans (worm) | 6.5 |
| Drosophila melanogaster (fly) | 3.1 |
| Takifugu rubripes (tiger pufferflsh) | 2.7 |
| Homo sapiens (human) | 44.4 |
Protein Diversity
In spite of only a modest increase in gene number, vertebrates have considerably more protein diversity than do invertebrates. One way to measure the amount of protein diversity is by counting the number of protein domains, which are characteristic parts of proteins that are often associated with a function. Vertebrate genomes do not encode significantly more protein domains than do invertebrate genomes; for example, there are 1262 domains in humans compared with 1035 in fruit flies (Table 20.4). However, the existing domains in humans are assembled into more combinations, leading to many more types of proteins.
| Species | Number of Predicted Protein Domains |
|---|---|
| Saccharomyces cerevisiae (yeast) | 851 |
| Arabidopsis thaliana (plant) | 1012 |
| Caenorhabditis elegans (roundworm) | 1014 |
| Drosophila melanogaster (fruit fly) | 1035 |
| Homo sapiens (human) | 1262 |
Source: Number of genes and protein-domain families from the International Human Genome Sequencing Consortium, Initial sequencing and analysis of the human genome, Nature 409:860-921, Table 23, 2001.
Homologous Genes
An obvious and remarkable trend seen in eukaryotic genomes is the degree of homology among genes found in even distantly related species. For example, mice and humans have about 99% of their genes in common. About 50% of the genes in fruit flies are homologous to genes in humans, and, even in plants, about 18% of the genes are homologous to those found in humans.
Collinearity Between Related Genomes
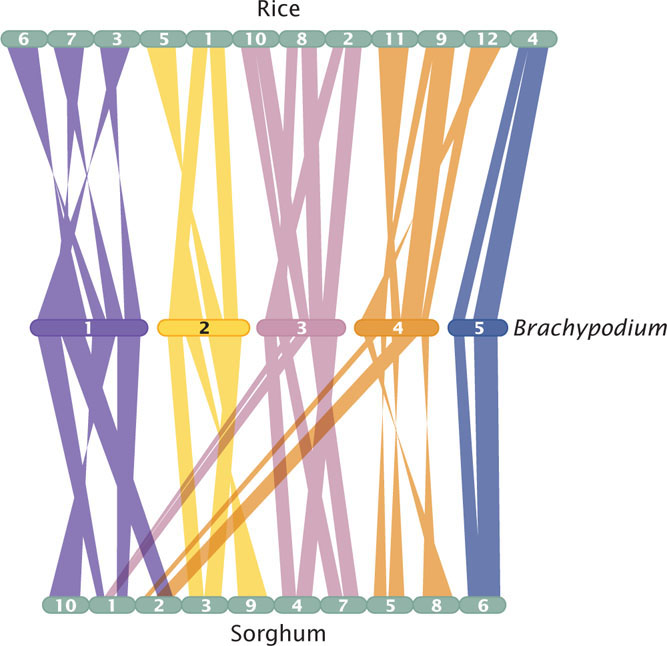
One of the features of genome evolution revealed by comparing the gene sequences of different organisms is that many genes are present in the same order in related genomes, a phenomenon that is sometimes termed collinearity. The reason for collinearity among genomes is that they are descended from a common ancestral genome, and evolutionary forces have maintained the same gene order in the genomes of descendants. Genomic studies of grasses—plants in the family Poaceae—illustrate the principle of collinearity.
Grasses comprise more than 10,000 species, including economically important crops such as rice, wheat, barley, corn, millet, oat, and sorghum. Taken together, grasses make up about 60% of the world’s food production. The genomes of these species vary greatly in size and chromosome number. For example, chromosome number in grasses ranges from 4 to 266; the rice genome consists of only about 460 million base pairs, whereas the genome of wheat contains 17 billion base pairs. In spite of these large differences in chromosome number and genome size, the position and order of many genes within the genomes are remarkably conserved. Through evolutionary time, regions of DNA between the genes (intergenic regions) have increased, decreased, and undergone rearrangements, whereas the genes themselves have stayed relatively constant in order and content. An example of the collinear relationship of genes between rice, sorghum, and Brachypodium (wild grass) is shown in Figure 20.15. ![]() TRY PROBLEM 38
TRY PROBLEM 38
601
CONCEPTS
Genome size varies greatly among eukaryotic species. For multicellular eukaryotic organisms, there is no clear relation between organismal complexity and amount of DNA or gene number. A substantial part of the genome in eukaryotic organisms consists of repetitive DNA, much of which is derived from transposable elements. Many eukaryotic genomes have homologous genes in common, and genes are often in the same order in the genomes of related organisms.
CONCEPT CHECK 8
Segmental duplications play an important role in evolution by
- giving rise to new genes and multigene families.
- keeping the number of genes in a genome constant.
- eliminating repetitive sequences produced by transposition.
- controlling the base content of the genome.
Comparative Drosophila Genomics
The fruit fly Drosophila melanogaster is one of the workhorses of genetics. Its genome was sequenced in 2000, the second animal genome to be deciphered. In 2007, researchers published genomic sequences of 10 additional species of Drosophila. Combined with the already sequenced genomes of D. melanogaster and D. pseudoobscura, this effort provided genomic data for 12 species of Drosophila, allowing detailed evolutionary analysis of this group of insects.
The 12 Drosophila species that were sequenced are found throughout the world, and all diverged from a common ancestor some 60 million years ago (Figure 20.16). Genome size in the group varies from 130 million base pairs in D. mojavensis to 364 million base pairs in D. virilis. The number of protein-encoding genes varies less widely, from 13,733 to 17,325. Many of the genes are found in all 12 of the species; for example, 77% of the 13,733 genes found in D. melanogaster have homologs in all of the other species.
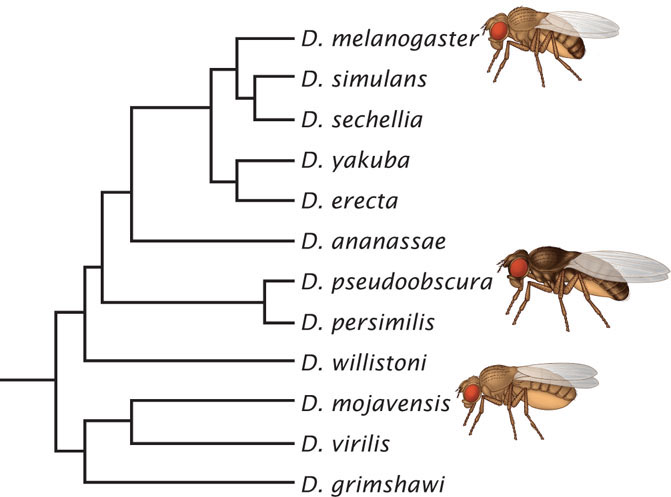
Transposable elements have played an important role in the evolution of Drosophila genomes. The extent of DNA that consists of the remnants of transposable elements ranges from 2.7% of the genome in D. simulans to 25% of the genome of D. ananassae. The amount of DNA contributed by transposable elements accounts for much of the difference in genome size among the species. Genomic rearrangements (inversions and translocations) were frequent in the evolution of this group. The rate of evolution varies among different classes of genes: genes controlling olfaction, immunity, and insecticide resistance have evolved at a faster rate compared with other genes.
The Human Genome
The human genome, which is fairly typical of mammalian genomes, has been extensively studied and analyzed because of its importance to human health and evolution. It is 3.2 billion base pairs in length, but only about 1.5% encodes proteins. Active genes are often separated by vast regions of noncoding DNA, much of which consists of repeated sequences derived from transposable elements.
602
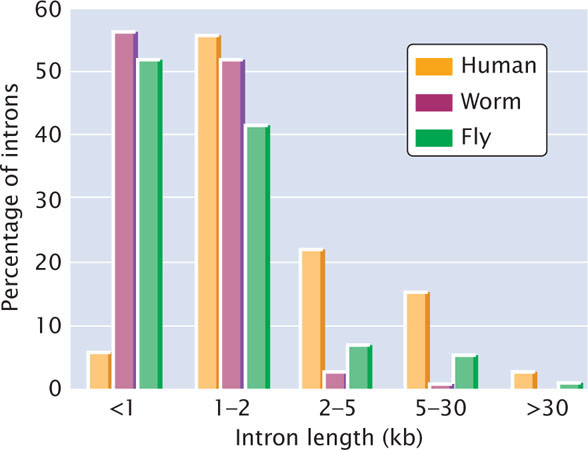
The average gene in the human genome is approximately 27,000 bp in length, with about 9 exons (Table 20.5). (One exceptional gene has 234 exons.) The introns of human genes are much longer, and there are more of them than in other genomes (Figure 20.17). The human genome does not encode substantially more protein domains (see Table 20.4), but the domains are combined in more ways to produce a relatively diverse proteome. Gene functions encoded by the human genome are presented in Figure 20.18. A single gene often encodes multiple proteins through alternative splicing; each gene encodes, on the average, two or three different mRNAs.
| Characteristic | Average |
|---|---|
| Number of exons | 8.8 |
| Size of internal exon | 145 bp |
| Size of intron | 3,365 bp |
| Size of 5′ untranslated region | 300 bp |
| Size of 3′ untranslated region | 770 bp |
| Size of coding region | 1,340 bp |
| Total length of gene | 27,000 bp |

Gene density varies among human chromosomes; chromosomes 17, 19, and 22 have the highest densities and chromosomes X, Y, 4, 13, and 18 have the lowest densities. Some proteins encoded by the human genome that are not found in other animals include: those affecting immune function; neural development, structure, and function; intercellular and intracellular signaling pathways in development; hemostasis; and apoptosis.
Transposable elements are much more common in the human genome than in worm, plant, and fruit-fly genomes (see Table 20.3). The human genome contains a variety of types of transposable elements, including LINEs, SINEs, retrotransposons, and DNA transposons (see Chapter 18). Most appear to be evolutionarily old and are defective, containing mutations and deletions making them no longer capable of transposition.
603