21.3 Epigenetic Processes Produce a Diverse Set of Effects
Initially, epigenetic mechanisms were thought to play a role in a small number of unusual phenotypes. However, research during the past 15 years has revealed that epigenetics underlies an impressive and ever-increasing array of biological phenomena. In this section, we will look at some examples of these epigenetic effects.
Paramutation
One of the first examples of epigenetics was a curious phenotype that Alexander Brink described in corn in the 1950s. Brink was studying the r1 locus, which helps to determine pigmentation in the seeds of corn. The Rr allele at this locus normally produces purple kernels, while the Rst allele codes for spotted kernels. Brink observed that when Rst was present in a genotype with Rr allele, the Rst allele permanently altered the expression of the Rr allele, so that it now also produced spotted seeds. This diminished effect of the altered Rr allele on pigmentation persisted for several generations, even in the absence of the Rst allele. Brink called this phenomenon paramutation. Paramutation violates Mendel’s principle of segregation, which states that when gametes are formed, each allele separates and is transmitted independently to the next generation.
Today, paramutation is defined as an interaction between two alleles that leads to a heritable change in expression of one of the alleles. Surprisingly, paramutation produces these differences in phenotype without any alteration in the DNA base sequence of the converted allele. The phenomenon of paramutation has several important features. First, the newly established expression pattern of the converted allele is transmitted to future generations, even though the allele that brought about the alteration is no longer present with it. Second, the altered allele is now able to convert other alleles to the new phenotype. And third, there are no associated DNA sequence differences in the altered alleles. A number of examples of paramutation have now been discovered in different organisms, and geneticists have begun to unravel the molecular mechanism of this curious phenomenon.
Paramutation in Corn
A few years after Brink reported paramutation at the rl locus in corn, another related example was discovered by Ed Coe, Jr. This case involved interaction between the alleles at the b1 locus in corn, which also aids in determining pigmentation. Paramutation at the b1 locus is more straightforward than at the rl locus, so we will use b1 to examine the process and mechanism of paramutation.
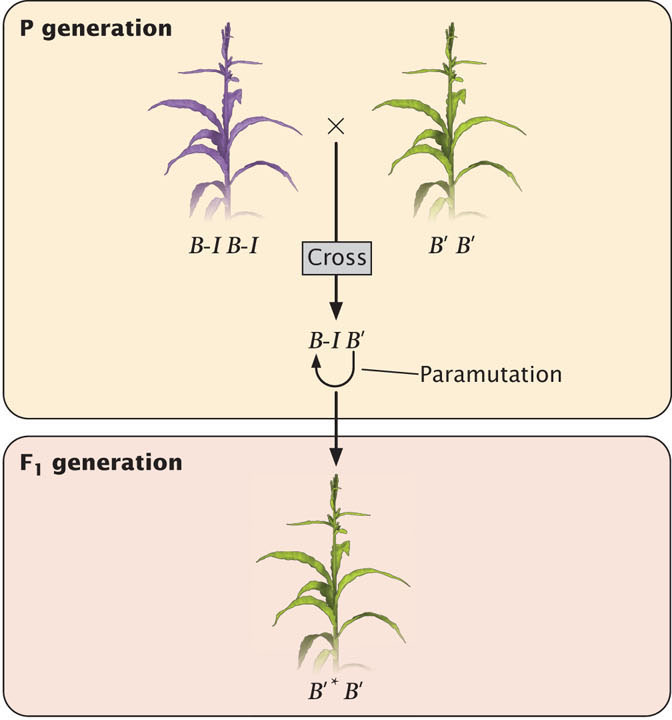
The b1 locus helps to determine the amount of purple anthocyanin that a corn plant produces. The locus actually encodes a transcription factor that regulates genes involved in pigment production. Plants homozygous for the B-I allele (B-I B-I) show high expression of the b1 locus and are dark purple (Figure 21.5). Plants homozygous for the B′ allele (B′B′) have a lower expression of b1 and are lightly pigmented. However, the DNA sequences of the B-I and B′ alleles are identical. Genetically identical alleles such as these, which produce heritable differences in phenotypes through epigenetic processes, are referred to as epialleles.
620
In plants that are heterozygous B-I B′, the B-I allele is converted to B′, with the result that the heterozygous plants are lightly pigmented (see Figure 21.5), just like the B′B′ homozygotes. The newly converted allele is usually designated B′*. Importantly, there is no functional difference between B' and B′*; the B′* allele is now fully capable of converting other B-I alleles into B′* alleles in subsequent generations.
Research has demonstrated that one of the features required for paramutation at the b1 locus is the presence of a series of seven tandemly repeated sequences that are located approximately 100,000 base pairs upstream of the coding sequence for the b1 locus (Figure 21.6). Each repeat consists of 853 bp and does not encode any protein. Both the B-I and B′ alleles have these tandem repeats, but the chromatin structure of the two alleles differs: the B-I allele has more open chromatin. The tandem repeats are required for high expression of the B-I allele and pigment production. It has been suggested that the repeats act like an enhancer (see Chapter 17), stimulating transcription at the b1 locus, but only when the chromatin surrounding the repeats is in an open configuration, as it is in the B-I allele. The more closed configuration of the B′ allele may prevent the repeats from interacting with the promoter of b1 and stimulating transcription. How the repeats might interact with the B′ allele is not known.
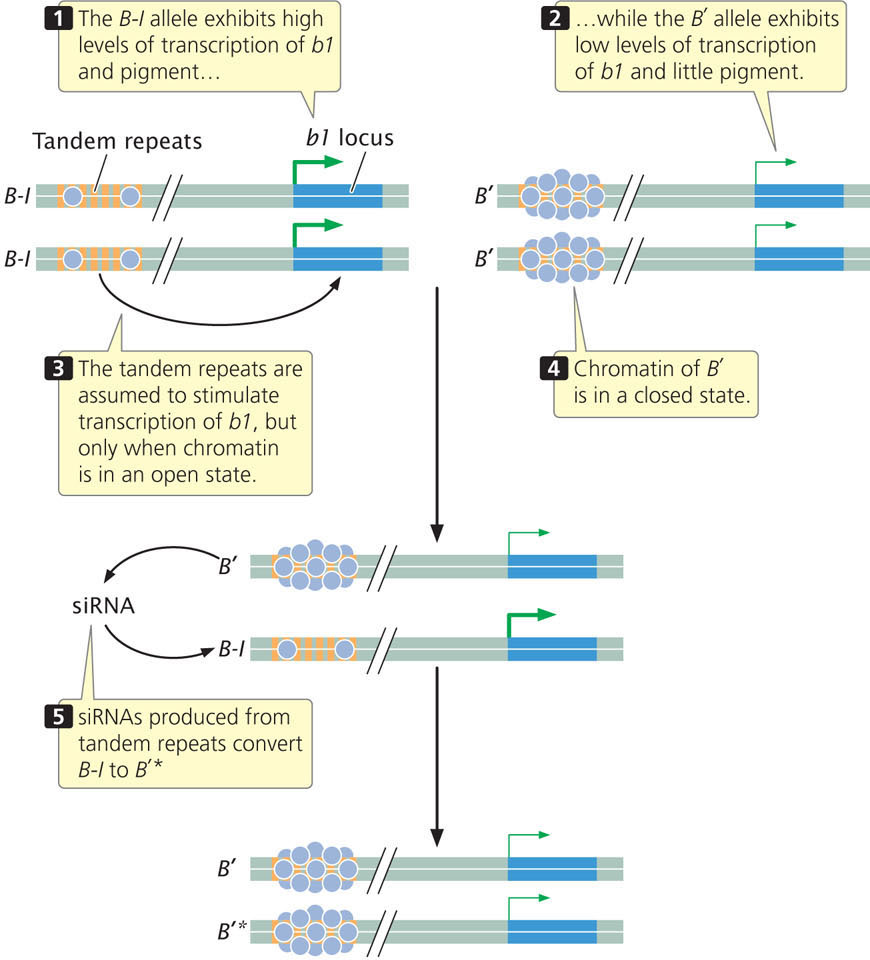
The different chromatin states of B-I and B′ may explain their different levels of expression, but how does the B′ allele convert the B-I allele to B′*? Although the mechanism is not completely understood, recent research demonstrates that the communication between B′ and B-I probably occurs through the action of small RNA molecules.
The tandem repeats that are required for paramutation encode 25-nucleotide long siRNAs, (Chapter 14). Some siRNAs are known to modify chromatin structure by directing DNA methylation to specific DNA sequences. Geneticists have isolated several genes in corn that are required for paramutation to take place; inactivating these genes eliminates paramutation. One of these genes is mop1, which encodes an RNA-directed RNA polymerase (an enzyme that synthesizes RNA from an RNA template). This gene is required to generate the siRNAs encoded by the tandem repeats, although it does not appear to be the enzyme that actually transcribes the DNA copies of tandem repeats. Another gene required for paramutation, called rmrl, encodes a chromatin-remodeling protein.
Thus, the current evidence suggests that siRNA molecules convert B-I to B′*, and this conversion probably involves a change to the chromatin states of the alleles. There are other examples from plants where siRNAs influence DNA methylation and chromatin structure. However, exactly how the siRNA molecules bring about this change in chromatin in paramutation is unknown. The altered chromatin state of the repeats in the B′ allele probably decreases transcription of the b1 locus, perhaps by interfering with interaction between the tandem repeats and the promoter of b1. It is assumed that the alteration to the chromatin of the B′ allele is stable and transmitted to future generations. Research also shows that the transcription of the tandem repeats and generation of siRNAs from them are necessary but not sufficient for paramutation, so additional factors must be involved. It is also not clear how the production of the siRNAs is transmitted across generations.
Paramutation in Mice
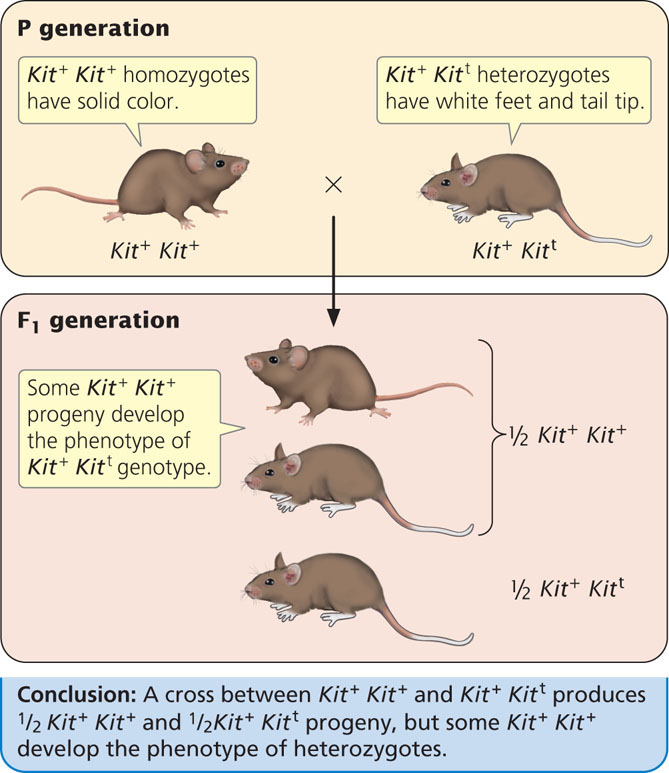
Several examples of paramutation have also been observed in mice. One involves the Kit locus, which encodes a tyrosine kinase receptor and functions in pigment production, germ cell development, and production of blood cells. Geneticists had earlier genetically engineered a mutant Kit allele (designated here as Kitt), which carries a 3000 bp portion of the lacZ gene (see Chapter 16) inserted into the Kit locus. Mice that are homozygous for the wild-type allele (Kit+ Kit+) have normal pigment. Mice that are homozygous for mutant Kit alleles (Kitt Kitt) die shortly after birth. Mice heterozygous for wild type and mutant alleles (Kit+ Kitt) have white tail tips and white feet (Figure 21.7). When a heterozygous mouse is crossed with a homozygous wild-type mouse, half of the progeny are homozygous (Kit+ Kit+) and half are heterozygous (Kit+ Kitt), as expected. However, many of the Kit+ Kit+ mice develop white tails and feet, the phenotype expected of the heterozygotes. In the presence of the Kitt allele in the heterozygote, the Kit+ allele is altered so that it has the same phenotype as Kitt. Mice with these altered alleles are designated as Kit*. The altered Kit* allele is stably transmitted to future generations, where it continues to produce white tails and feet. Some humans with a white spot in the forehead hair and areas of reduced pigment (called the piebald trait) have mutations in the Kit locus; other mutations in Kit result in a predisposition to some cancers.
621
Researchers have demonstrated that this example of paramutation is also mediated through RNA molecules, although the mechanism is likely to be different from that seen in paramutation in corn. In mice, the white tail and feet of the Kitt allele appears to be caused by microRNAs (miRNAs) that degrade the Kit mRNA, and these miRNAs are transmitted to future generations through the gametes. Researchers observed a two-fold decrease in Kit mRNA in both the heterozygous mice and the Kit* mice, suggesting that the white tails and feet of the heterozygotes is due to a reduction in the amount of Kit mRNA. To determine whether RNA was responsible for paramutation they injected some wild-type embryos with RNA from Kit+ Kit+ homozygotes and injected other wild-type embryos with RNA from Kit+ Kitt heterozygotes. Among the mice that completed development, they observed white tail tips and feet more frequently in those injected with RNA from heterozygotes, suggesting that RNA from the heterozygotes is capable of altering the Kit+ allele of the wild type mice (Table 21.1). The researchers then injected into wild-type embryos miRNAs that degrade Kit mRNA. This produced more mice with white tails and feet than when they injected nonspecific miRNAs into the embryos (Table 21.1) The ability to produce the white tails and feet characteristic of the Kit+ Kitt genotype by injecting miRNA into embryos suggests that this case of paramutation is associated with miRNA molecules that are transferred to the embryo via egg and sperm. However, there are still many unknown aspects of paramutation at the Kit locus.
| Type of RNA Injected | Presence of White Tail Tips and Feet |
|---|---|
| Kit+ Kit+ mRNA | Uncommon |
| Kit+ Kitt mRNA | More common |
| imiRNA to Kit mRNA | More common |
| nonspecific miRNA | Uncommon |
CONCEPTS
Paramutation occurs when one allele creates a heritable alteration of another allele without any change in DNA sequence. Research suggests that paramutation in corn and mice is mediated through small RNA molecules.
 CONCEPT CHECK 3
CONCEPT CHECK 3
Which is a characteristic of paramutation?
- One allele is able to alter another allele when both are present in a heterozygote.
- Altered alleles must be passed on to future generations.
- Altered alleles must be capable of altering other alleles in future generations.
- All of the above.
Behavioral Epigenetics
Research has shown that life experiences, especially those early in life, can have long-lasting effects on behavior, in some cases into future generations. Increasingly, researchers are finding that these long-term effects are mediated through epigenetic processes. The number of studies that convincingly demonstrate that life experience alters chromatin structure is currently small (and some are still controversial), but a number of researchers are actively looking for epigenetic effects of experience and their long-term effects on chromatin structure and behavior.
622
Epigenetic Changes Induced by Maternal Behavior
A fascinating example of behavioral epigenetics is seen in the long-lasting effects of maternal behavior in rats. A mother rat licks and grooms her offspring (Figure 21.8), usually while she arches her back and nurses them. The offspring of mothers who display more licking and grooming behavior are less fearful as adults and show reduced hormonal responses to stress compared with the offspring of mothers who lick and groom less. These long-lasting differences in the offspring are not due to genetic differences inherited from their mothers—at least not genetic differences in the base sequences of the DNA. Offspring exposed to more licking and grooming develop a different pattern of DNA methylation compared with offspring exposed to less licking and grooming. These differences in DNA methylation affect the acetylation of histone proteins that persist into adulthood and alter the expression of the glucocorticoid receptor gene, which plays a role in hormonal responses to stress. The expression of other stress-response genes also is affected.

To demonstrate the effect of altered chromatin structure on the stress response of the offspring, researchers infused the brains of young rats with a deacetylase inhibitor, which prevents the removal of acetyl groups from the histone proteins. After infusion of the deacetylase inhibitor, differences in DNA methylation and histone acetylation associated with grooming behavior disappeared, as did the difference in responses to fear and stress in the adults. This demonstrates that the mother rat’s licking and grooming behavior brings about epigenetic changes in the offspring’s chromatin, which causes long-lasting differences in their behavior. ![]() TRY PROBLEM 28
TRY PROBLEM 28
Epigenetic Effects of Early Stress in Humans
Numerous studies have demonstrated that stress during childhood and adolescence produce a number of adverse effects that persist into adult life. For example, childhood abuse increases the probability that the child will experience depression, anxiety, and suicide as an adult. In one study, researchers examined the brains of 24 people who had committed suicide, half of whom had experienced childhood abuse. They found that those who experienced childhood abuse had a greater degree of methylation of the glucocorticoid receptor gene, a gene involved in the stress response, than those who had not experienced abuse. Although the number of brains studied was small, the study suggests that early childhood stress can indeed cause epigenetic modifications to chromatin structure in humans.
Other studies have demonstrated that gene expression is affected by early life experience. For example, researchers found that growing up in a lower socioeconomic environment before the age of 5 altered the expression of over 100 genes related to immune function of adults. The introduction to Chapter 11 discusses the observation that early childhood stress—in the form of growing up in an orphanage—alters telomere length, a type of epigenetic change.
Epigenetics in Cognition
A number of research studies have shown that abnormalities in DNA methylation are associated with disorders of development and intellectual ability in humans. These findings prompted researchers to look for effects of chromatin structure on learning, memory, and cognitive ability in mice and rats. One study found that training mice to avoid an adverse stimulus at a specific location reduced DNA methylation of the Bdnf gene, which encodes a growth factor that stimulates the growth of connections between neurons. When demethylated, the Bdnf gene was more active. When researchers injected into the mice’s brains a drug that inhibits demethylation, activity of the Bdnf gene was decreased, and the mice’s memory of where the adverse stimulus occurred also decreased.
Another study found that a drug that promotes the acetylation of histone proteins improved learning and memory in mice that have a disorder similar to Alzheimer disease. Acetylation of histones alters chromatin structure by loosening the association of DNA and histone proteins and stimulates transcription of many genes. Other studies have found that histone acetylation decreases with age in mice, with diminished expression of genes related to learning and memory. When researchers injected mice with a drug that is an inhibitor of deacetylase activity, acetylation of histones increased, transcription of genes involved in memory increased, and memory of the mice improved. These studies suggest that changes in chromatin structure may be involved in memory and learning.
CONCEPTS
Studies are providing evidence that early life experiences can produce epigenetic changes that have long-lasting effects on behavior.
623
Epigenetic Effects of Environmental Chemicals
Because some chemicals are capable of modifying chromatin structure, researchers have looked for long-term effects of environmental toxicants on chromatin structure and epigenetic traits.
There has been much recent interest in chemicals, called endocrine disruptors, which mimic or interfere with natural hormones. Endocrine disruptors are capable of interfering with processes regulated by natural hormones, such as sexual development and reproduction. One endocrine disruptor is vinclozolin, a common fungicide used to control fungal diseases in grapes, fruits, and vegetables and to treat turf on golf courses. Vinclozolin acts as an antagonist at the androgen receptor—vinclozolin and its metabolites mimic testosterone and bind to the androgen receptor, preventing testosterone from binding. But vinclozolin and its metabolites do not properly activate the receptor and, in this way, vinclozolin inhibits the action of androgens and prevents sperm production.
In one study, researchers found that the exposure of embryonic rats to vinclozolin led to reduced sperm production not only in the treated animals (when they reached puberty), but also in several subsequent generations. Increased DNA methylation was seen in sperm of the males that were exposed to vinclozolin and these patterns of methylation were inherited. This study and others have raised concerns that, through epigenetic changes, environmental exposure to some chemicals might have effects on the health of future generations. ![]() TRY PROBLEM 28
TRY PROBLEM 28
CONCEPTS
Through epigenetic changes, environmental chemicals may have influences that extend to later generations.
Transgenerational Epigenetic Effects on Metabolism
In the introduction to this chapter, we discussed how diet during childhood can have effects on health that can carry across generations. These types of epidemiological studies on humans are supported by laboratory studies of mice and rats. In one study, researchers fed inbred male mice either a normal (control) diet or a diet low in protein. They then bred mice in both groups to control females fed a normal diet. The males were then separated from the females and never had any contact with their offspring; their only contribution to the offspring was a set of paternal genes transferred through the sperm.
The offspring were raised and their lipid and cholesterol levels examined. The offspring of males fed a low protein diet exhibited increased expression of genes involved in lipid and cholesterol metabolism and a corresponding decrease in levels of cholesterol, compared to the offspring of males fed a normal diet. They also observed numerous differences in DNA methylation in the offspring of the two types of fathers, although no differences could be found in the methylation patterns of the sperm of the two groups of fathers. These results suggest that epigenetic changes altered the cholesterol metabolism of the offspring, although how the differences in methylation were transmitted from father to offspring was unclear.
In another study, researchers fed male rats a high-fat diet and, not surprisingly, they gained weight. They then bred these males to females that had been fed a normal diet. The offspring were also fed a normal diet. The daughters of the male rats on the high-fat diet had normal weight, but as adults they developed a diabetes-like condition of impaired glucose tolerance and insulin secretion. The researchers observed that in the insulin-secreting pancreatic islet cells of the daughters the expression of 642 genes involved in insulin secretion and glucose tolerance was altered, demonstrating that the father’s diet affected gene expression in his daughters.
Epigenetic Effects in Monozygotic Twins
Monozygotic (identical) twins develop from a single egg fertilized by a single sperm that divides and gives rise to two zygotes (see Chapter 6). Monozygotic twins are genetically identical, in the sense that they possess identical DNA sequences, but they often differ somewhat in appearance, health, and behavior. The nature of these differences in the phenotypes of identical twins is not well understood, but recent evidence suggests that at least some of these differences may be due to epigenetic changes. In one study, Mario Fraga at the Spanish National Cancer Center and his colleagues examined 80 identical twins and compared the degree and location of their DNA methylation and histone acetylation. They found that DNA methylation and histone acetylation in identical twins were similar early in life, but older twins had remarkable differences in their overall content and distribution of DNA methylation and histone acetylation. Furthermore, these differences affected gene expression in the twins. This research suggests that identical twins do differ epigenetically and that phenotypic differences between them may be caused by differential gene expression.
CONCEPTS
Phenotypic differences between genetically identical monozygotic twins may result from epigenetic effects.
CONCEPT CHECK 4
What degree of differences would you expect to see in the DNA base sequences and epigenetic marks of monozygotic twins?
- Similar differences in DNA base sequence and epigenetic marks.
- Greater differences in DNA base sequence than epigenetic marks.
- Greater differences in epigenetic marks than DNA base sequence.
- No differences in either DNA base sequence or epigenetic marks.
X Inactivation
In female mammals, one X chromosome in each cell is randomly inactivated to provide equal expression of X-linked genes in males and females (see Chapter 4). Through this process, termed X inactivation, many genes on the inactivated X chromosome are permanently silenced and are not transcribed. Once a particular X chromosome is inactivated in a cell, that same X chromosome remains inactivated when the DNA is replicated, and the inactivation mark is passed on to daughter cells through mitosis. This phenomenon is responsible for the patchy distribution of black and orange pigment seen in tortoiseshell cats (see Chapter 4). X inactivation is a type of epigenetic effect because it results in a stable change in gene expression that is passed on to other cells.
624
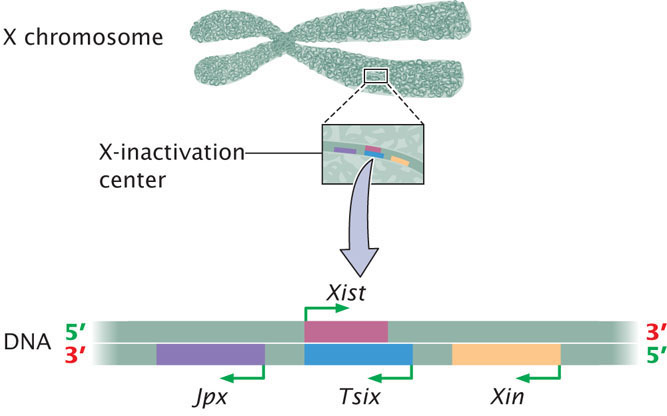
A great deal of research has demonstrated that which X chromosome is inactivated within a cell is controlled by a particular segment of the X chromosome called the X-inactivation center, which is 100,000 to 500,000 bp in length. Inactivation is initiated at the X-inactivation center and then spreads to the remainder of the inactivated X chromosome. Examination of the X-inactivation center led to the discovery of several genes that play a role in inactivating one X chromosome in each female cell and keeping the other X chromosome active (Figure 21.9).
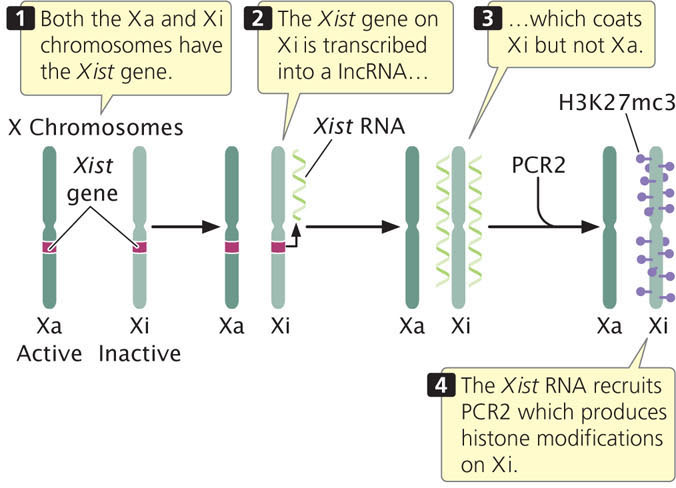
The key player in X inactivation is a gene called Xist (for X-inactivation-specific transcript) which encodes a long noncoding RNA (lncRNA) that is 17,000 bp in length (Figure 21.10). As its name implies, this RNA molecule does not encode a protein. Instead, Xist lncRNA coats the X chromosome from which it was transcribed. Xist lncRNA then attracts polycomb repressor complex 2 (PRC2) and eventually polycomb repressor complex 1 (PRC1). These proteins produce epigenetic marks, such as histone 3 lysine 27 trimethylation (H3K27me3) and other histone modifications that repress transcription. Eventually, many CpG dinucleotides are methylated, leading to permanent silencing of the inactivated X chromosome.
In mice, there are two separate inactivation events. Soon after fertilization, when the embryo reaches the 8-cell stage, the X chromosome from the male parent is inactivated, while the maternal X chromosome remains active. In the developing embryo, the paternal X chromosome is then reactivated during blastocyst maturation. Inactivation occurs again in early development, but now which X is inactivated is random—the X from the male parent and the X from the female parent are equally likely to be inactivated. From this point on, whichever X is inactivated remains silenced through subsequent cell divisions. However, some genes on the inactivated X chromosome escape inactivation and continue to be transcribed. How these genes escape X inactivation is not known. Interestingly, in marsupial mammals, the paternal X chromosome is the copy that remains permanently silenced in all cells.
As mentioned, X-inactivation is brought about by the transcription of the Xist gene on the inactive X chromosome to produce Xist lncRNA, which coats the inactive X chromosome and leads to changes in chromatin structure that silence transcription. But what happens on the active X chromosome? Why isn’t it coated by Xist RNA and silenced? Although all details of this process are not yet understood, recent research has demonstrated that there are several additional genes in the X-inactivation center that encode other IncRNAs. These lncRNAs help bring about X inactivation of the inactive X, while not silencing the active X (see Figure 21.9). One of these is the Tsix gene, which is transcribed on the active X chromosome. Tsix is antisense to Xist, which means that it overlaps with the Xist gene and is transcribed from the opposite strand (see Figure 21.10), producing a Tsix IncRNA that is complementary to Xist lncRNA. Through several mechanisms, Tsix represses the expression of Xist on the active X chromosome. Another major player is a gene called Jpx, which encodes an lncRNA that stimulates transcription of Xist on the inactive X chromosome. Thus, Xist is controlled by two parallel switches with opposite effects: (1) Jpx stimulates Xist expression on the inactive X chromosome, causing Xist to be transcribed and leading to X-inactivation; and (2) Tsix represses Xist on the active X chromosome, causing Xist not to be transcribed on that chromosome and preventing inactivation. Several other genes are also involved. A gene called Xite encodes a lncRNA that sustains Tsix expression on the active X chromosome. The major genes involved in the process of X-inactivation are summarized in Table 21.2.
| Gene | Encodes | Action of Gene |
|---|---|---|
| Xist | lncRNA | Coats inactive X chromosome and leads to silencing of transcription of many genes on the inactive X |
| Tsix | lncRNA | Inhibits transcription of Xist on active X chromosome |
| Jpx | lncRNA | Stimulates transcription of Xist on inactive X chromosome |
| Xite | lncRNA | Sustains Tsix expression on active X, which inhibits Xist and maintains transcription of genes on active X chromosome |
625
This complex process ensures that in each female cell one X chromosome is inactivated and one remains active. Scientists have long recognized that X-inactivation also involves some type of mechanism that is capable of counting X chromosomes, because all but one X chromosome in each cell is inactivated. Thus, the single X in the cells of an XY male remains active (no X-inactivation occurs), and two X chromosomes are inactivated in XXX females (see discussion of Barr bodies in Chapter 4). The nature of this counting mechanism is not yet well understood. ![]() TRY PROBLEM 31
TRY PROBLEM 31
CONCEPTS
Epigenetic changes underlie X-inactivation, in which one X chromosome in female cells is permanently silenced. X-inactivation occurs through the action of several genes in the X-inactivation center that encode long noncoding RNAs. The products of these genes interact to ensure that one X chromosome is inactive and one remains active in each female cell.
CONCEPT CHECK 5
What would be the effect of introducing into a female cell siRNAs that degrade Xist RNA?
Epigenetic Changes Associated with Cell Differentiation
All cells in the human body are genetically identical, and yet different cell types exhibit remarkably different phenotypes—a nerve cell is quite distinct in its shape, size, and function from an intestinal cell. These differences in phenotypes are stable and passed from one cell to another, despite the fact that the DNA sequences of all the cells are the same.
Stem cells are undifferentiated cells that are capable of forming every type of cell in an organism, a property referred to as pluripotency. As a stem cell divides and gives rise to a more specialized type of cell, the gene-expression program of the cell becomes progressively fixed, so that each particular cell type expresses only those genes necessary to carry out the functions of that cell type. Though the control of these cell-specific expression programs is not well understood, changes in DNA methylation and chromatin structure clearly play important roles in silencing some genes and activating others.
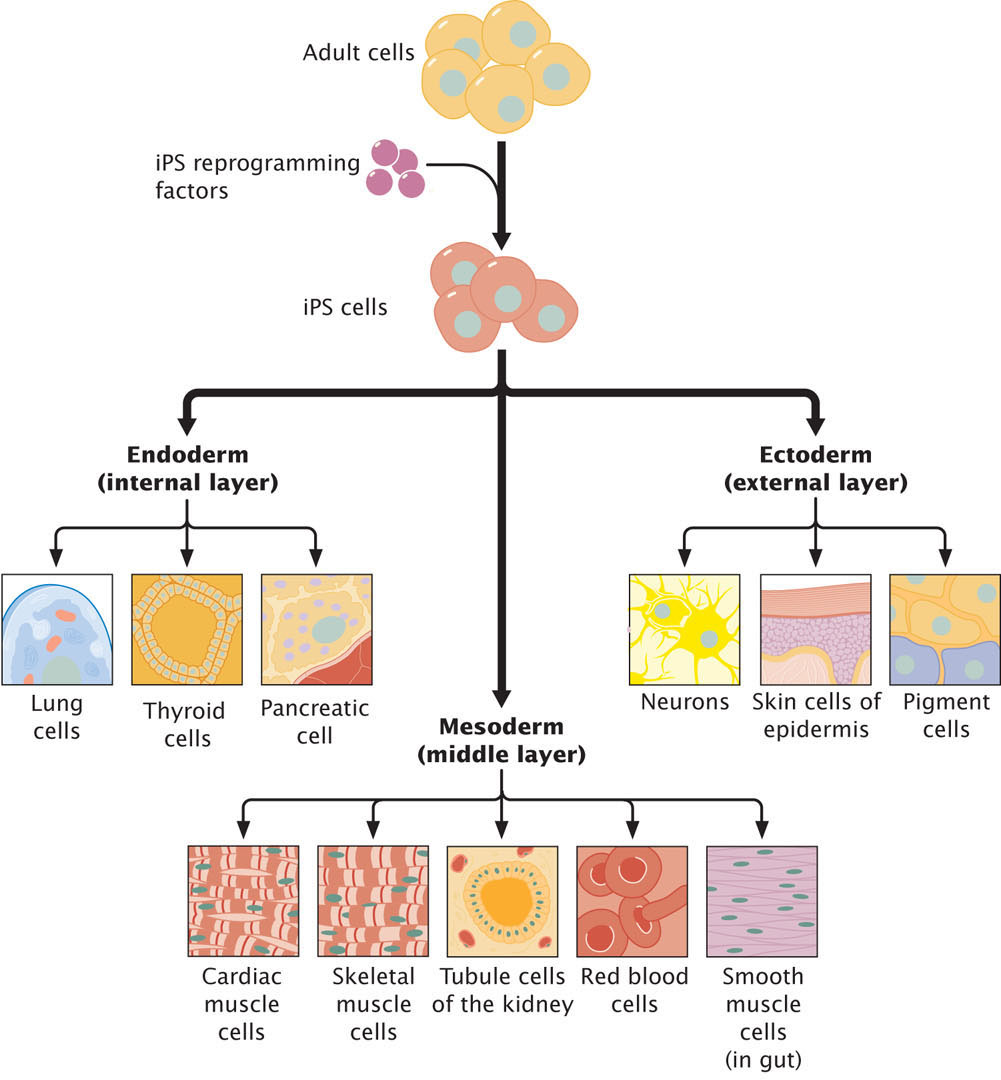
Stem cells provide a potential source of cells for regeneration of tissues, medical treatment, and research. In the past, the only source of stem cells with the capacity to differentiation into adult tissues were cells from embryos, but because of ethical concerns about creating and using human embryos for harvesting stem cells, researchers have long sought the ability to induce adult somatic cells to dedifferentiate and revert to stem cells. Such cells are called induced pluripotent stem cells (iPSCs). Researchers have now successfully created iPSCs by treating fibroblasts (fully differentiated human connective tissue cells) in culture with a cocktail of transcription factors (Figure 21.11), although less than 1% of the cells that are treated actually revert to iPSCs. Transcription factors that induce pluripotency cause extensive epigenetic reprogramming, altering patterns of DNA methylation and histone modifications that accumulate with cellular differentiation. Recent research has shown, however, that iPSCs retain a memory of their past and are not completely equivalent to embryonic stem cells (those derived from embryos). One study found that although the DNA methylation patterns of iPSCs differ greatly from those of differentiated somatic cells, the iPSCs retained some methylation marks of the somatic cells and that the methylation of iPSCs was not identical with that of embryonic stem cells. Another study compared histone modifications of fibroblasts, iPSCs, and embryonic stem cells. The iPSCs and embryonic stem cells had many fewer H3K27me3 and H3K9me3 marks than did the fibroblasts, but researchers also found significantly more of these marks on iPSCs than on embryonic stem cells.
626
Genomic Imprinting
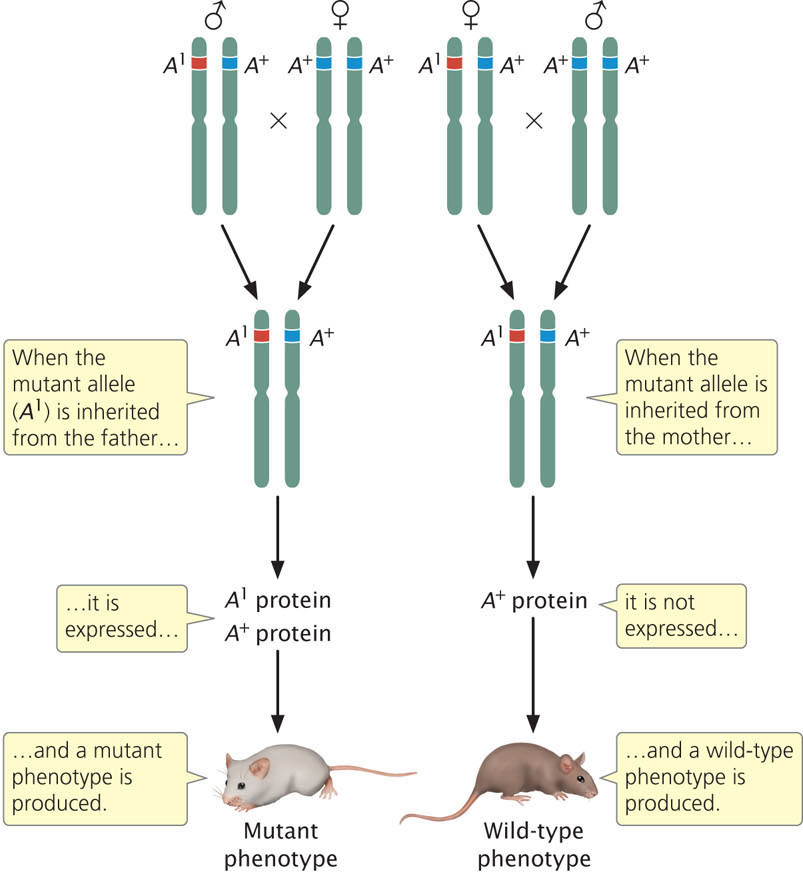
Diploid organisms usually possess two alleles at each autosomal locus, one allele inherited from the mother and one allele inherited from the father. For most genes, both alleles are expressed, and the effect of a particular allele on the phenotype is independent of which parent transmitted the allele to the offspring. However, for a few genes, the sex of the parent that contributed the allele influences how the allele is expressed—alleles inherited from the mother and father are not equivalent (Figure 21.12). This phenomenon, in which the sex of the parent that transmits the allele determines its expression, is termed genomic imprinting (see Chapter 5). For some imprinted genes, the allele inherited from the male parent is expressed and the allele inherited from the female parent is silent; for other genes, the allele inherited from the female parent is expressed and the allele inherited from the male parent is silent. As discussed in Chapter 5, genomic imprinting is thought to be due to different degrees of methylation of genes inherited from the parents.
Previous research suggested that the number of imprinted genes was limited, but more recent research suggests the number is much higher. A study conducted by Christopher Gregg at Harvard University and his colleagues found that over 1300 genes in the mouse brain exhibit evidence of genomic imprinting. Many of these imprinted genes were not completely silenced: there was biased expression, with one sex transmitting an allele that was more highly expressed than the allele transmitted by the other sex. Gregg and his colleagues also found that imprinting was highly variable; some genes were imprinted only in certain tissues or at certain times of development.
Genomic imprinting has a number of interesting parallels to X-inactivation. Most imprinted genes are located in clusters of 3-12 genes that occur at a discrete region of a particular chromosome. Each cluster contains genes that encode proteins, as well as genes that produce noncoding RNA. In each of the well-studied examples, there is an imprinting control region that determines imprinting; deletion of this region destroys the ability to imprint. In addition, the imprinting control region exhibits different chromatin modifications between alleles inherited from the male and female parents. Each imprinting cluster contains genes for one or more lncRNAs that play an important role in imprinting and are themselves imprinted. For example, the gene for insulin-like growth factor 2 (Igf2) in humans exhibits genomic imprinting; the Igf2 allele transmitted from the male parent is expressed, while the Igf2 allele transmitted from the female is silenced (see Figure 5.18 in Chapter 5). Several lncRNAs produced by other genes in the imprinting control region are required for gene silencing of Igf2 in females, although how they bring about repression of transcription is not clear.
Many of the well-studied clusters of imprinted genes are associated with disorders that result from faulty imprinting. Beckwith–Wiedemann syndrome is one such disorder. Children with Beckwith–Wiedemann syndrome exhibit excessive growth during fetal development and early childhood. They also have unusual embryonic malignant tumors. Beckwith–Wiedemann syndrome is associated with imprinting of a cluster of genes on chromosome 11, including the Igf2 gene. Individuals with Beckwith–Wiedemann syndrome often have small deletions on chromosome 11 that interfere with the normal process of imprinting. For example, Igf2 is normally expressed only when inherited from the father, but in some children with Beckwith–Wiedemann syndrome, deletions within the imprinting center lead to expression of alleles from both parents. The result is that too much Igf2 is produced, leading to excessive growth and cancer. Prader–Willi syndrome and Angelman syndrome are disorders that are due to imprinting defects on chromosome 15 (see Chapter 5).
627
Imprinting and Genetic Conflict
Many genes that are genomically imprinted affect fetal and early embryonic growth. One possible explanation for genomic imprinting is the genetic-conflict hypothesis, which suggests that there are different and conflicting evolutionary pressures acting on maternal and paternal alleles for genes (such as Igf2) that affect fetal growth. From an evolutionary standpoint, paternal alleles that maximize the size of the offspring are favored, because birth weight is strongly associated with infant mortality and adult health. Thus, it is to the advantage of the male parent to pass on alleles that promote maximum fetal growth of their offspring. In contrast, maternal alleles that cause more-limited fetal growth are favored: committing too many nutrients to any one fetus may limit a mother’s ability to reproduce in the future and giving birth to very large babies is also difficult and risky. The genetic-conflict hypothesis predicts that genomic imprinting will evolve: paternal copies of genes that affect fetal growth should be maximally expressed, whereas maternal copies of the same genes should be less actively expressed or even silent. Indeed, Igf2 follows this pattern: the paternal allele is active and promotes growth; the maternal allele is silent and does not contribute to growth. Recent findings demonstrate that the paternal copy of Igf2 promotes fetal growth by directing more maternal nutrients to the fetus through the placenta.
CONCEPTS
Genomic imprinting is caused by epigenetic differences in the alleles inherited from male and female parents. The genetic conflict hypothesis suggests that imprinting evolves because of conflicting evolutionary pressures acting on maternal and paternal alleles.
CONCEPT CHECK 6
Which is true of genomic imprinting?
- The sex of the parent that transmits an allele affects the expression of the allele in the offspring.
- The sex of the offspring affects the expression of an allele inherited from one of the parents.
- The sex of the parent affects how an allele is transmitted to the offspring.
- The sex of the offspring affects which allele is inherited from the parent.