23.2 Mutations in a Number of Different Types of Genes Contribute to Cancer
As we have learned, cancer is a disease caused by alterations in DNA. However, there are many different types of genetic alterations that may contribute to cancer. More than 350 different human genes have been identified that contribute to cancer; the actual number is probably much higher. Research on mice suggests that more than 2000 genes can, when mutated, contribute to the development of cancer. In the next several sections, we will consider some of the different types of genes that frequently have roles in cancer.
Oncogenes and Tumor-Suppressor Genes
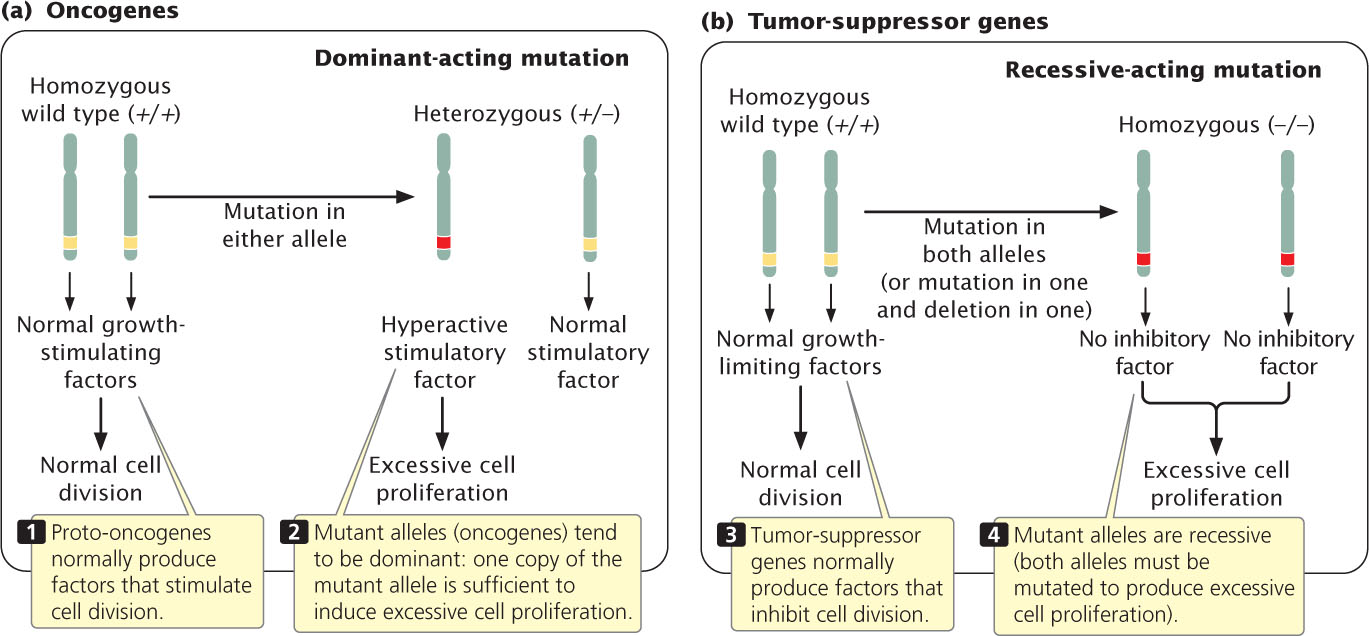
The signals that regulate cell division fall into two basic types: molecules that stimulate cell division and those that inhibit it. These control mechanisms are similar to the accelerator and brake of a car. In normal cells (but, one would hope, not your car), both accelerators and brakes are applied at the same time, causing cell division to proceed at the proper speed.
Because cell division is affected by both accelerators and brakes, cancer can arise from mutations in either type of signal, and there are several fundamentally different routes to cancer. A stimulatory gene can be made hyperactive or active at inappropriate times, analogous to having a car’s accelerator stuck in the floored position. Mutations in stimulatory genes are usually dominant because even the reduced amount of gene product produced by a single allele is usually sufficient to produce a stimulatory effect. Mutated dominant-acting stimulatory genes that cause cancer are termed oncogenes (Figure 23.5a). Cell division may also be stimulated when inhibitory genes are made inactive, analogous to having a defective brake in a car. Mutated inhibitory genes generally have recessive effects, because both copies must be mutated to remove all inhibition. Inhibitory genes in cancer are termed tumor-suppressor genes (Figure 23.5b). Many cancer cells have mutations in both oncogenes and tumor-suppressor genes.
667
Although oncogenes or mutated tumor-suppressor genes or both are required to produce cancer, mutations in DNA-repair genes can increase the likelihood of acquiring mutations in these genes. Having mutated DNA-repair genes is analogous to having a lousy car mechanic who does not make the necessary repairs on a broken accelerator or brake.
Oncogenes
Oncogenes were the first cancer-causing genes to be identified. In 1909, a farmer brought physician Peyton Rous a hen with a large connective-tissue tumor (sarcoma) growing on its breast. When Rous injected pieces of this tumor into other hens, they also developed sarcomas. Rous conducted experiments that demonstrated that the tumors were being transmitted by a virus, which became known as the Rous sarcoma virus. A number of other cancer-causing viruses were subsequently isolated from various animal tissues. These viruses were generally assumed to carry a cancer-causing gene that was transferred to the host cell. The first oncogene, called src, was isolated from the Rous sarcoma virus in 1970.
In 1975, Michael Bishop, Harold Varmus, and their colleagues began to use probes for viral oncogenes to search for related sequences in normal cells. They discovered that the genomes of all normal cells carry DNA sequences that are closely related to oncogenes. These normal cellular genes are called proto-oncogenes. They are responsible for basic cellular functions in normal cells but, when mutated, become oncogenes that contribute to the development of cancer. When a virus infects a cell, a proto-oncogene may become incorporated into the viral genome through recombination. Within the viral genome, the proto-oncogene may mutate to an oncogene that, when inserted back into a cell, causes rapid cell division and cancer. Because the proto-oncogenes are more likely to undergo mutation or recombination within a virus, viral infection is often associated with the cancer.
Proto-oncogenes can be converted into oncogenes in viruses several different ways. The sequence of the proto-oncogene may be altered or truncated as it is incorporated into the viral genome. This mutated copy of the gene may then produce an altered protein that causes uncontrolled cell proliferation. Alternatively, through recombination a proto-oncogene may end up next to a viral promoter or enhancer, which then causes the gene to be overexpressed. Finally, sometimes the function of a proto-oncogene in the host cell may be altered when a virus inserts its own DNA into the gene, disrupting its normal function. While viruses are capable of converting proto-oncogenes into oncogenes, most proto-oncogenes are mutated to form oncogenes without the involvement of a virus.
Many oncogenes have been identified by experiments in which selected fragments of DNA are added to cells in culture. Some of the cells take up the DNA and, if these cells become cancerous, then the DNA fragment that was added to the culture must contain an oncogene. The fragments can then be sequenced, and the oncogene can be identified. A large number of oncogenes have now been discovered (Table 23.4). About 90% of all cancer genes are thought to be dominant oncogenes.
| Gene | Normal function | Cancer in which gene is mutated |
|---|---|---|
| erbB | Part of growth factor receptor | Many types of cancer |
| fos | Transcription factor | Osteosarcoma and endometrial carcinoma |
| jun | Transcription factor, cell cycle control | Lung cancer, breast cancer |
| myc | Transcription factor | Lymphomas, leukemias, neuroblastoma |
| ras | GTp binding and GTpase | Many types of cancer |
| sis | Growth factor | Glioblastomas and other cancers |
| src | Protein tyrosine kinase | Many types of cancer |
Tumor-Suppressor Genes
Tumor-suppressor genes are more difficult to identify than oncogenes because they inhibit cancer and are recessive; both alleles must be mutated before the inhibition of cell division is removed. Because the failure of their function promotes cell proliferation, tumor-suppressor genes cannot be identified by adding them to cells and looking for cancer. About 10% of cancer-causing genes are thought to be tumor-suppressor genes.
Defects in both copies of a tumor-suppressor gene are usually required to cause cancer; an organism can inherit one defective copy of the tumor-suppressor gene (is heterozygous for the cancer-causing mutation) and not have cancer, because the remaining normal allele produces the tumor-suppressing product. However, these heterozygotes are often predisposed to cancer because the inactivation or loss of the one remaining allele is all that is required to completely eliminate the tumor-suppressor product. Inactivation of the remaining wild-type allele in heterozygotes is referred to as the loss of heterozygosity. A common mechanism for the loss of heterozygosity is a deletion on the chromosome that carried the normal copy of the tumor-suppressor gene (Figure 23.6).
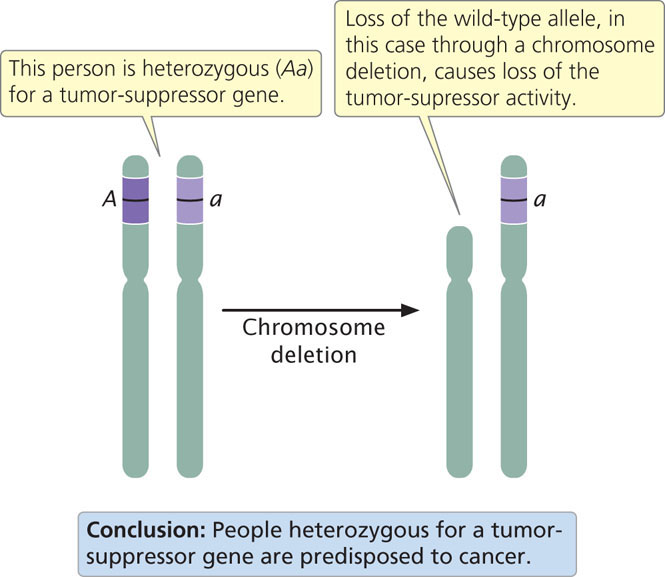
Among the first tumor-suppressor genes to be identified was the gene causing retinoblastoma. In 1985, Raymond White and Webster Cavenne showed that large segments of chromosome 13 were missing in cells of retinoblastoma tumors, and, later, the tumor-suppressor gene was isolated from these segments. Another example of a tumor-suppressor gene is BRCA1, mutations of which are associated with increased risk of breast and ovarian cancer. BRCA1 produces a protein that normally helps in repair of double-strand breaks in DNA by homologous recombination (see Chapter 18). It also acts as a transcription factor and interacts with histone deacetylase enzymes, which affect transcription. A number of tumor-suppressor genes have now been discovered (Table 23.5).
| Gene | Normal function | Cancer in which gene is mutated |
|---|---|---|
| APC | Scaffold protein, interacts with microtubules | Colorectal |
| BRCA1 | DNA repair, transcription factor | Breast and ovarian |
| CDKN2A | Regulates cell division | Melanoma |
| NF1 | GTpase activator | Neurofibromatosis |
| p53 | Regulates cell division | Many types of cancer |
| RB | Regulates cell division | Retinoblastoma |
668
Sometimes the mutation or loss of a single allele of a recessive tumor-suppressor gene is sufficient to cause cancer. This effect—the appearance of the trait in an individual cell or organism that is heterozygous for a normally recessive trait—is called haploinsufficiency. This phenomenon is thought to be due to dosage effects: the heterozygote produces only half as much of the product encoded by the tumor-suppressing gene. Normally, this amount is sufficient for the cellular processes that prevent tumor formation, but it is less than the optimal amount, and other factors may sometimes combine with the lowered tumor-suppressor product to cause cancer. ![]() TRY PROBLEM 25
TRY PROBLEM 25
CONCEPTS
Proto-oncogenes are genes that control normal cellular functions; when mutated, they become oncogenes that stimulate cell proliferation. They tend to be dominant in their action. Tumor-suppressor genes normally inhibit cell proliferation; when mutated, they allow cells to proliferate. Tumor-suppressor genes tend to be recessive in their action. Individual organisms that are heterozygous for tumor-suppressor genes are often predisposed to cancer.
 CONCEPT CHECK 2
CONCEPT CHECK 2
Why are oncogenes usually dominant in their action, whereas tumor-suppressor genes are recessive?
Mutations in Genes That Control the Cycle of Cell Division
The cell cycle is the normal process by which cells undergo growth and division. Normally, progression through the cell cycle is tightly regulated so that cells divide only when additional cells are needed, when all the components necessary for division are present, and when the DNA has been replicated without damage. Sometimes, however, errors arise in one or more of the components that regulate the cell cycle. These errors often cause cells to divide at inappropriate times or rates, leading to cancer. Indeed, many proto-oncogenes and tumor-suppressor genes function normally by helping to control the cell cycle. Before considering how errors in this system contribute to cancer, we must first understand how the cell cycle is usually regulated.
Control of the Cell Cycle
As discussed in Chapter 2, the cell cycle is the period from one cell division to the next. Cells that are actively dividing pass through the G1, S, and G2 phases of interphase and then move directly into the M phase, when cell division takes place. Nondividing cells exit from G1 into the G0 stage, in which they are functional but not actively growing or dividing. Progression from one stage of the cell cycle to another is influenced by a number of internal and external signals and is regulated at key points in the cycle called checkpoints.
For many years, the biochemical events that control the progression of cells through the cell cycle were completely unknown, but research has now revealed many of the details of this process. Key events of the cell cycle are controlled by cyclin-dependent kinases (CDKs), which are enzymes that add phosphate groups to other proteins. Sometimes phosphorylation activates the other protein and other times it inactivates the protein. As their name implies, CDKs are functional only when they associate with another protein called a cyclin. The level of cyclin oscillates in the course of the cell cycle; when bound to a CDK, cyclin specifies which proteins the CDK will phosphorylate. Each cyclin appears at a specific point in the cell cycle, usually because its synthesis and destruction are regulated by another cyclin. Cyclins and CDKs are called by different names in different organisms; here, we will use the terms applied to these molecules in mammals.
669
G1-TO-S Transition
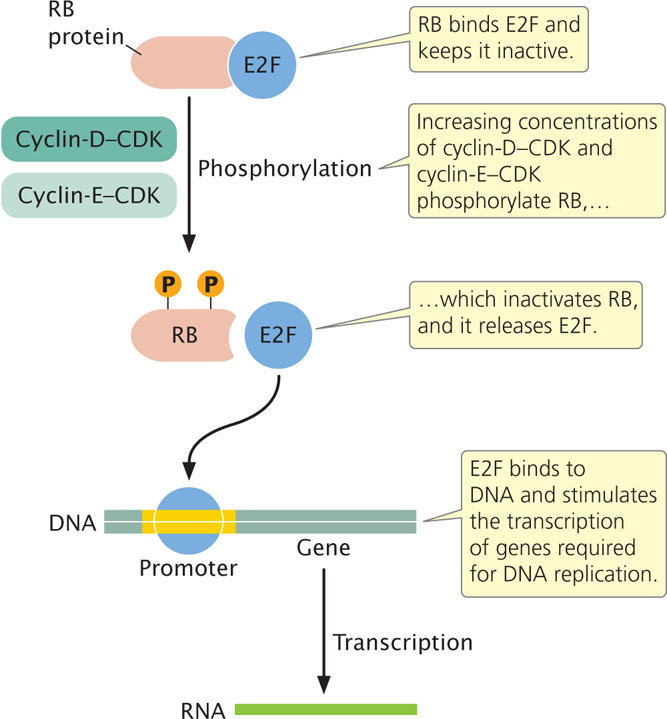
Let’s begin by looking at the G1-to-S transition. As mentioned earlier, checkpoints ensure that all cellular components are present and in good working order before the cell proceeds to the next stage of the cycle. The G1/S checkpoint is in G1, just before the cell enters into the S phase and replicates its DNA. The cell is prevented from passing through the G1/S checkpoint by the retinoblastoma (RB) protein (Figure 23.7), which binds to another molecule called E2F and keeps it inactive. In G1, cyclin D and cyclin E continuously increase in concentration and combine with their associated CDKs. Cyclin-D-CDK and cyclin-E-CDK both phosphorylate molecules of RB. Late in G1, the phosphorylation of RB is completed, which inactivates RB. Without the inhibitory effects of RB, the E2F protein is released. E2F is a transcription factor that stimulates the transcription of genes that produce enzymes necessary for the replication of DNA, and the cell moves into the S stage of the cell cycle.
G2-TO-M Transition
Regulation of the G2-to-M transition is similar to that of the G1-to-S transition. In the G2-to-M transition, cyclin B combines with a CDK to form an inactive complex called mitosis-promoting factor (MPF). After MPF has been formed, it must be activated by the removal of a phosphate group (Figure 23.8a). During G1, cyclin B levels are low; so the amount of MPF also is low. As more cyclin B is produced, it combines with CDK to form increasing amounts of MPF. Near the end of G2, the amount of active MPF reaches a critical level, which commits the cell to divide. The MPF concentration continues to increase, reaching a peak in mitosis.
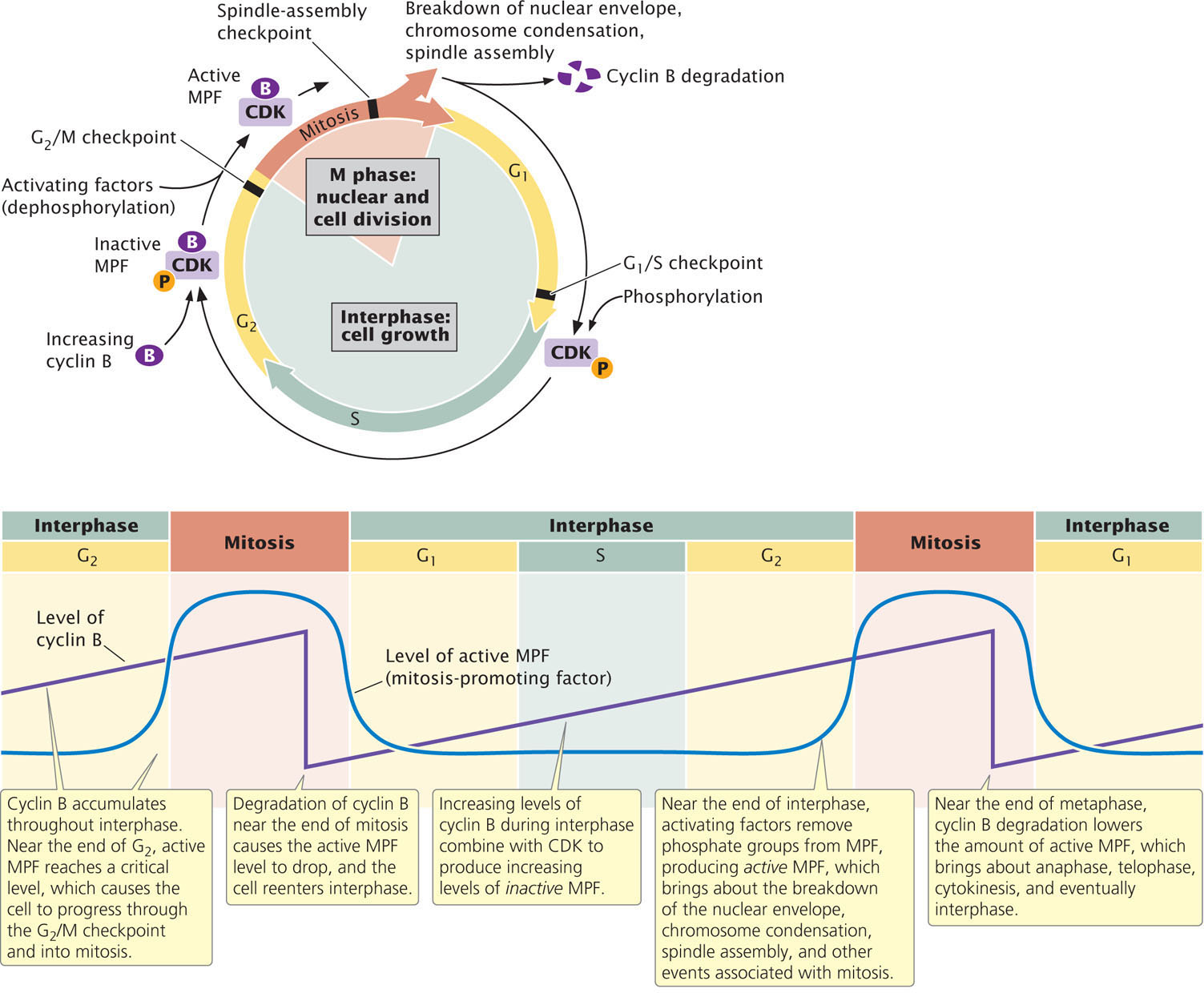
The active form of MPF phosphorylates other proteins, which then bring about many of the events associated with mitosis, such as nuclear-membrane breakdown, spindle formation, and chromosome condensation. At the end of metaphase, cyclin B is abruptly degraded, which lowers the amount of MPF and, initiating anaphase, sets in motion a chain of events that ultimately brings mitosis to a close (Figure 23.8b). In brief, high levels of active MPF stimulate mitosis, and low levels of MPF bring a return to interphase conditions.
The G2/M checkpoint is at the end of G2, before the cell enters mitosis. A number of factors stimulate the synthesis of cyclin B and the activation of MPF, whereas other factors inhibit MPF. Together, these factors ensure that mitosis is not initiated until conditions are appropriate for cell division. For example, DNA damage inhibits the activation of MPF; consequently, the cell is arrested in G2 and does not undergo division.
Spindle-Assembly Checkpoint
Yet another checkpoint, called the spindle-assembly checkpoint, is in metaphase. This checkpoint delays the onset of anaphase until all chromosomes are aligned on the metaphase plate and sister kinetochores are attached to spindle fibers from opposite poles. If all chromosomes are not properly aligned, the checkpoint blocks the destruction of cyclin B. The persistence of cyclin B keeps MPF active and maintains the cell in a mitotic state. An additional checkpoint controls the cell’s exit from mitosis.
Mutations in Cell-Cycle Control and Cancer
Many cancers are caused by defects in the cell cycle’s regulatory machinery. For example, mutations in the gene that encodes the RB protein—which normally holds the cell in G1 until the DNA is ready to be replicated—are associated with many cancers, including retinoblastoma. When the RB gene is mutated, cells pass through the G1/S checkpoint without the normal controls that prevent cell proliferation. The gene that encodes cyclin D (thus stimulating the passage of cells through the G1/S checkpoint) is overexpressed in about 50% of all breast cancers, as well as some cases of esophageal and skin cancer. Likewise, the tumor-suppressor gene p53, which is mutated in about 75% of all colon cancers, regulates a potent inhibitor of CDK activity.
Some proto-oncogenes and tumor-suppressor genes have roles in apoptosis, a process of programmed cell death in which the cell’s DNA is degraded, its nucleus and cytoplasm shrink, and the cell undergoes phagocytosis by other cells without the leakage of its contents. Cells have the ability to assess themselves and, when they are abnormal or damaged, they normally undergo apoptosis. Cancer cells frequently have chromosome mutations, DNA damage, and other cellular anomalies that would normally stimulate apoptosis and prevent their proliferation. Often, these cells have mutations in genes that regulate apoptosis, and therefore they do not undergo programmed cell death. The ability of a cell to initiate apoptosis in response to DNA damage, for example, depends on p53, which is inactive in many human cancers.
670
CONCEPTS
Progression through the cell cycle is controlled at checkpoints, which are regulated by interactions between cyclins and cyclin-dependent kinases. Genes that control the cell cycle are frequently mutated in cancer cells.
CONCEPT CHECK 3
What would be the most likely effect of a mutation that causes cyclin B to be unable to bind to CDK?
- Cells pass through the G2/M checkpoint and enter mitosis even when DNA has not been replicated.
- Cells never pass through the G1/S checkpoint.
- Cells pass through mitosis more quickly than unmutated cells.
- Cells fail to pass the G2/M checkpoint and do not enter into mitosis.
Signal-Transduction Pathways
Whether cells pass through the cell cycle and continue to divide is influenced by a large number of internal and external signals. External signals are initiated by hormones and growth factors. These molecules are often unable to pass through the cell membrane because of their size or charge; they exert their effects by binding to receptors on the cell surface, which triggers a series of intracellular reactions that then carry the message to the nucleus or other site within the cell. This type of system, in which an external signal triggers a cascade of intracellular reactions that ultimately produce a specific response, is called a signal-transduction pathway. Defects in signal-transduction pathways are often associated with cancer.
671
A signal-transduction pathway begins with the binding of an external signaling molecule to a specific receptor that is embedded in the cell membrane. Receptors in signal-transduction pathways usually have three parts: (1) an extracellular domain that protrudes from the cell and binds the signaling molecule; (2) a transmembrane domain that passes across the membrane and conducts the signal to the interior of the cell; and (3) an intracellular domain that extends into the cytoplasm and, on the binding of the signaling molecule, undergoes a chemical or conformational change that is transmitted to molecules of the signal-transduction pathway in the cytoplasm. The binding of a signaling molecule to the membrane-bound receptor activates a protein in the pathway. On activation, this protein activates the next molecule in the pathway, often by adding or removing phosphate groups or causing changes in the conformation of the protein. The newly activated protein activates the next molecule in the pathway, and in this way, the signal is passed along through a cascade of reactions and ultimately produces the response, such as stimulating or inhibiting the cell cycle.
In the past decade, much research has been conducted to determine the pathways by which various signals influence the cell cycle. To illustrate signal transduction, let’s consider the Ras signal-transduction pathway, which plays an important role in control of the cell cycle. Each Ras protein cycles between an active form and an inactive form. In the inactive form, the Ras protein is bound to guanosine diphosphate (GDP); in the active form, it is bound to guanosine triphosphate (GTP).
The Ras signal-transduction pathway is activated when a growth factor, such as epidermal growth factor (EGF), binds to a receptor on the cell membrane (Figure 23.9). The binding of EGF causes a conformational change in the receptor and the addition of phosphate groups to it. The addition of the phosphate groups allows adaptor molecules to bind to the receptor. These adaptor molecules link the receptor with an inactive molecule of Ras protein. The adaptor molecules stimulate Ras to release GDP and bind GTP, activating Ras. The newly activated Ras protein then binds to an inactive form of another protein called Raf and activates it. After activating Raf, Ras hydrolyzes GTP to GDP, which converts Ras back into the inactive form.
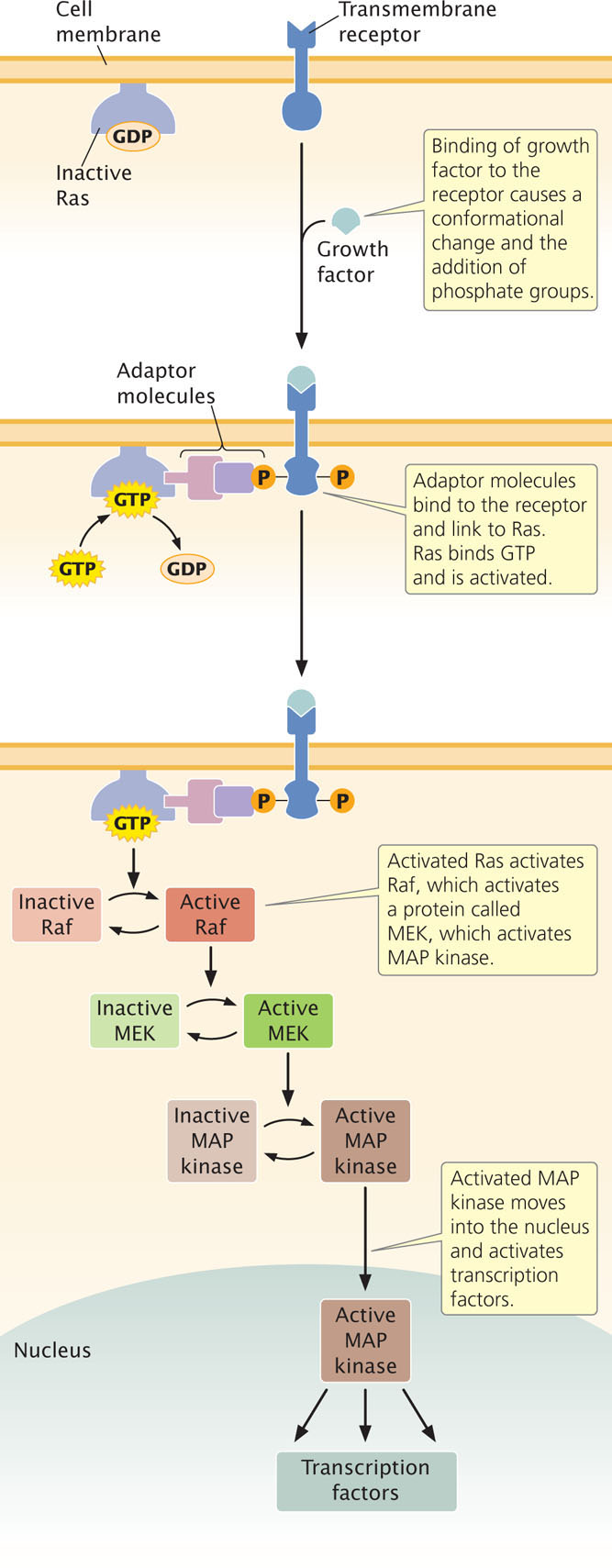
Activated Raf then sets in motion a cascade of reactions, ending in the activation of a protein called MAP kinase. Activated MAP kinase moves into the nucleus and activates a number of transcription factors that stimulate the transcription of genes taking part in the cell cycle. In this way, the original external signal promotes cell division. A number of other transduction pathways have been identified that affect the cell cycle and cell proliferation.
672
Because signal-transduction pathways help control the cell cycle, defects in their components often contribute to cancer. For example, genes that encode Ras proteins are frequently oncogenes, and mutations in these genes are often found in cancer cells: 75% of tumors of the pancreas and 50% of those in the thyroid and colon have mutations in ras genes. Mutations in these genes produce mutant Ras proteins that are permanently activated and continuously stimulate cell division.
CONCEPTS
Molecules outside the cell often bring about intracellular responses by binding to a membrane receptor and stimulating a cascade of intracellular reactions, known as a signal-transduction pathway. Many molecules in the pathway are proteins that alternate between active and inactive forms. Defects in signal-transduction pathways are often associated with cancer.
CONCEPT CHECK 4
Ras proteins are activated when they
- bind GTR
- release GTp.
- bind GDp.
- undergo acetylation.
DNA-Repair Genes
Cancer arises from the accumulation of multiple mutations in a single cell. Some cancer cells have normal rates of mutation, and multiple mutations accumulate because each mutation gives the cell a replicative advantage over cells without the mutations. Other cancer cells may have higher-than-normal rates of mutation in all of their genes, which leads to more-frequent mutation of oncogenes and tumor-suppressor genes. What might be the source of these high rates of mutation in some cancer cells?
Two processes control the rate at which mutations arise within a cell: (1) the rate at which errors arise during and after the course of replication and (2) the efficiency with which these errors are corrected. The error rate in replication is controlled by the fidelity of DNA polymerases and other proteins in the replication process (see Chapter 12). However, defects in genes encoding replication proteins have not been strongly linked to cancer.
The mutation rate is also strongly affected by whether errors are corrected by DNA-repair systems. Defects in genes that encode components of these repair systems have been consistently associated with a number of cancers. People with xeroderma pigmentosum, for example, are defective in nucleotide-excision repair, an important cellular repair system that normally corrects DNA damage caused by a number of mutagens, including ultraviolet light. Likewise, about 13% of colorectal, endometrial, and stomach cancers have cells that are defective in mismatch repair, another major repair system in the cell.
A particular type of colon cancer called nonpolyposis colorectal cancer is inherited as an autosomal dominant trait. In families with this condition, a person can inherit one mutated and one normal allele of a gene that controls mismatch repair. The normal allele provides sufficient levels of the protein for mismatch repair to function, but it is highly likely that this normal allele will become mutated or lost in at least a few cells. If it does so, there is no mismatch repair, and these cells undergo higher-than-normal rates of mutation, leading to defects in oncogenes and tumor-suppressor genes that cause the cells to proliferate.
Defects in DNA-repair systems may also contribute to the generation of chromosome rearrangements and genomic instability. Many DNA-repair systems make s ingle- and double-strand breaks in the DNA. If these breaks are not repaired properly, then chromosome rearrangements often result.
Genes That Regulate Telomerase
Another factor that may contribute to the progression of cancer is the inappropriate activation of the enzyme telomerase. Telomeres are special sequences at the ends of eukaryotic chromosomes. Recall that the ends of chromosomes cannot be replicated, and telomeres become shorter with each cell division. This shortening eventually leads to the destruction of the chromosome and cell death, so somatic cells are only capable of a limited number of cell divisions.
In germ cells and stem cells, telomerase replicates the chromosome ends, thereby maintaining the telomeres, but this enzyme is not normally expressed in somatic cells. In many tumor cells, however, sequences that regulate the expression of the telomerase gene are mutated, allowing the enzyme to be expressed, and the cell is capable of unlimited cell division. This mutation allows cancer cells to divide indefinitely. Although the expression of telomerase appears to contribute to the development of many cancers, its precise role in tumor progression is unknown and under investigation.
Genes That Promote Vascularization and the Spread of Tumors
A final set of factors that contribute to the progression of cancer includes genes that affect the growth and spread of tumors. Oxygen and nutrients, which are essential to the survival and growth of tumors, are supplied by blood vessels, and the growth of new blood vessels (angiogenesis) is important to tumor progression. Angiogenesis is stimulated by growth factors and others proteins encoded by genes whose expression is carefully regulated in normal cells. In tumor cells, genes encoding these proteins are often overexpressed compared with normal cells, and inhibitors of angiogenesis-promoting factors may be inactivated or underexpressed. At least one inherited cancer—von Hippel-Lindau disease, in which people develop multiple types of tumors—is caused by the mutation of a gene that affects angiogenesis.
673
In the development of many cancers, the primary tumor gives rise to cells that spread to distant sites, producing secondary tumors. This process of metastasis is the cause of death in 90% of human cancer cases; it is influenced by cellular changes induced by somatic mutation. As discussed in the introduction to this chapter, the palladin gene, when mutated, contributes to the metastasis of pancreatic tumors. By using microarrays to measure levels of gene expression, researchers have identified other genes that are transcribed at a significantly higher rate in metastatic cells compared with nonmetastatic cells. For example, one study detected a set of 95 genes that were overexpressed or underexpressed in a population of metastatic breast-cancer cells that were strongly metastatic to the lung, compared with a population of cells that were only weakly metastatic to the lung. Genes that contribute to metastasis often encode components of the extracellular matrix and the cytoskeleton. Others encode adhesion proteins, which help hold cells together.
Advances in sequencing technology have now made possible the complete sequencing of the DNA of tumor cells to see how their genomes differ from those of normal cells. In one experiment, researchers sequenced the entire genome of cells from a metastasized breast-cancer tumor and compared it with the genome of noncancer cells from the same person. They also compared the genome of the metastasized tumor with the genome of the primary tumor (from which the metastasis originated), which had been removed from the patient nine years earlier. The researchers found 32 different somatic mutations in the coding regions of genes from the tumor cells, 19 of which were not detected in the primary tumor. This finding suggests that the metastasized tumor underwent considerable genetic changes in its nine-year evolution from the primary tumor. In contrast, another study of a breast-cancer metastasis found only two mutations that were not present in the primary tumor but, in this case, the metastasis had evolved in only one year.
CONCEPTS
Mutations in genes that encode components of DNA-repair systems are often associated with cancer; these mutations increase the rate at which mutations are retained and result in an increased number of mutations in proto-oncogenes, tumor-suppressor genes, and other genes that contribute to cell proliferation. Mutations that allow telomerase to be expressed in somatic cells and those that affect vascularization and metastasis also can contribute to cancer progression.
CONCEPT CHECK 5
Which type of mutation in telomerase is associated with cancer cells?
- Mutations that produce an inactive form of telomerase.
- Mutations that decrease the expression of telomerase.
- Mutations that increase the expression of telomerase.
- All of the above.
MicroRNAs and Cancer
MicroRNAs (miRNAs) are a class of small RNA molecules that pair with complementary sequences on mRNA and degrade the mRNA or inhibit its translation (see Chapter 14). Given the fact that miRNAs are important in controlling gene expression and development, it is not surprising that they are also associated with tumor development. Many tumor cells exhibit widespread reduction in the expression of many miRNAs. Researchers have genetically engineered mouse tumor cells that lacked the machinery to generate miRNAs and found that these cells showed enhanced tumor progression when implanted into mice. Interestingly, this effect was seen only in cells that had already initiated tumor development, suggesting that miRNAs play a role in later stages of tumor progression.
Lowered levels of miRNAs may contribute to cancer by allowing oncogenes that are normally controlled by the miRNAs to be expressed at high levels. For example, let-7 miRNA normally controls the expression of the ras oncogene, probably by binding to complementary sequences in the 3′ untranslated region of mRNA and inhibiting translation. In lung-cancer cells, levels of let-7 miRNA are often low, allowing the Ras protein to be highly expressed, which then leads to the development of lung cancer.
A transcription factor called c-MYC is often expressed at high levels in cancer cells. Evidence suggests that c-MYC helps to drive cell proliferation and the development of cancer. Among other effects, c-MYC binds to the promoters of miRNA genes and decreases their transcription, decreasing the abundance of the miRNAs. Some of these miRNAs are known to suppress tumor development. Research has shown that, if, through genetic manipulation, the miRNAs are expressed at high levels, the development of tumors decreases. All of these findings suggest that altered expression of miRNAs plays an important role in cancer.
Several miRNAs have been implicated in the process of metastasis. A particular miRNA called miR-10b has been associated with the formation of metastatic breast tumors. In one experiment, investigators manipulated a line of breast-cancer cells so that miR-10b was overexpressed. When the manipulated cells were injected into mice, many of the mice developed metastatic tumors. In contrast with the preceding examples in which the lower expression of miRNA is associated with cancer, here high levels of miRNA appear to promote the spread of cancer cells. Further study revealed that, in humans, the levels of miR-10b are elevated in metastatic tumors compared with tumors in metastasis-free patients. miR-10b regulates the expression of a number of other genes, including some that are known to suppress the spread of tumor cells. Other miRNAs are known to inhibit metastasis.
674
Cancer Genome Projects
Formed in 2008, the International Cancer Genome Consortium coordinates efforts to determine the genomic sequences of tumors. A goal of the consortium is to completely sequence 500 tumors from each of 50 different types of cancer, along with the genomes of normal tissues from the same persons. This effort is producing important results, revealing the numbers and types of mutations that are associated with particular cancers. The hope is that new cancer-causing genes will be identified, which will lead to a better understanding of the nature of cancer and suggest new targets for cancer treatment. Another research project called The Cancer Genome Atlas (TCGA) project began in the United States in 2005. This project seeks to provide a comprehensive genomic analysis of over 100 different types of cancer cells, including sequencing of all exons across the genome, as well as characterization of mRNA expression, DNA methylation, copy number variations, and microRNAs of the tumor cells.
The genomes of a number of tumors have been sequenced and many more are currently being sequenced. For example, the entire genome of a small-cell lung carcinoma (a type of lung cancer) was sequenced in 2010 and compared with the genome of normal cells from the same person. More than 22,000 base-pair mutations were identified in the tumor, of which 134 were within protein-encoding genes. The tumor also possessed 58 chromosome rearrangements and 334 copy-number variations (see Chapter 20). In another study that was part of TCGA, researchers examined mRNA expression, microRNAs, DNA methylation, and copy number variations in 489 ovarian adenocarcinomas, and sequenced the DNA of exons from 316 of the tumors. Almost all of the tumors contained mutations in p53, a tumor-suppressor gene involved in DNA repair and cell cycle control. Mutations in BRCA1 and BRCA2, two tumor-suppressor genes that also occur in breast cancer, occurred in 22% of the tumors. Mutations in seven other genes occurred statistically more often than in normal cells. The results suggested several new approaches for drug treatment of ovarian cancer.
Another series of studies sequenced a number of genes in samples of malignant gliomas, an incurable and deadly form of brain cancer. The researchers examined DNA sequences, copy number variations, DNA methylation, and RNA expression of these tumors. The analyses revealed mutations in several genes that appear to be important in the development of glioma tumors. All of these genomic studies are providing new insight in the genetic basis of cancer.
The large number of mutations found in cancer genomes can be divided into two types: mutation drivers and mutation passengers. Drivers are mutations that drive the cancer process: they directly contribute to the development of cancer. Drivers include mutations in oncogenes, tumor-suppressor genes, DNA-repair genes, and the other types of cancer genes discussed in this chapter. Passengers are mutations that arise randomly in the process of tumor development and do not contribute to the cancer process. Many passengers are in introns (regions between genes) and other DNA that is not transcribed and translated, but they can also arise within protein-encoding genes. A major challenge is to determine which of the numerous mutations found in tumors are drivers and actually contribute to the development of cancer and which are passengers with no effect.