Concept 16.3: Phylogeny Makes Biology Comparative and Predictive
Once a phylogeny is reconstructed, what do we do with it? What beyond an understanding of evolutionary history does phylogeny offer us?
Phylogenies are important for reconstructing past events
Reconstructing past events is important for understanding many biological processes. In the case of zoonotic diseases (diseases caused by infectious organisms transmitted to humans from another animal host), it is important to understand when, where, and how the disease first entered a human population. Human immunodeficiency virus (HIV) is the cause of such a zoonotic disease, acquired immunodeficiency syndrome, or AIDS. Phylogenetic analyses have become important for studying the transmission of viruses such as HIV. Phylogenies are also important for understanding the present global diversity of HIV and for determining the virus’s origins in human populations. A broader phylogenetic analysis of immunodeficiency viruses shows that humans acquired these viruses from two different hosts: HIV-1 from chimpanzees, and HIV-2 from sooty mangabeys (FIGURE 16.6).
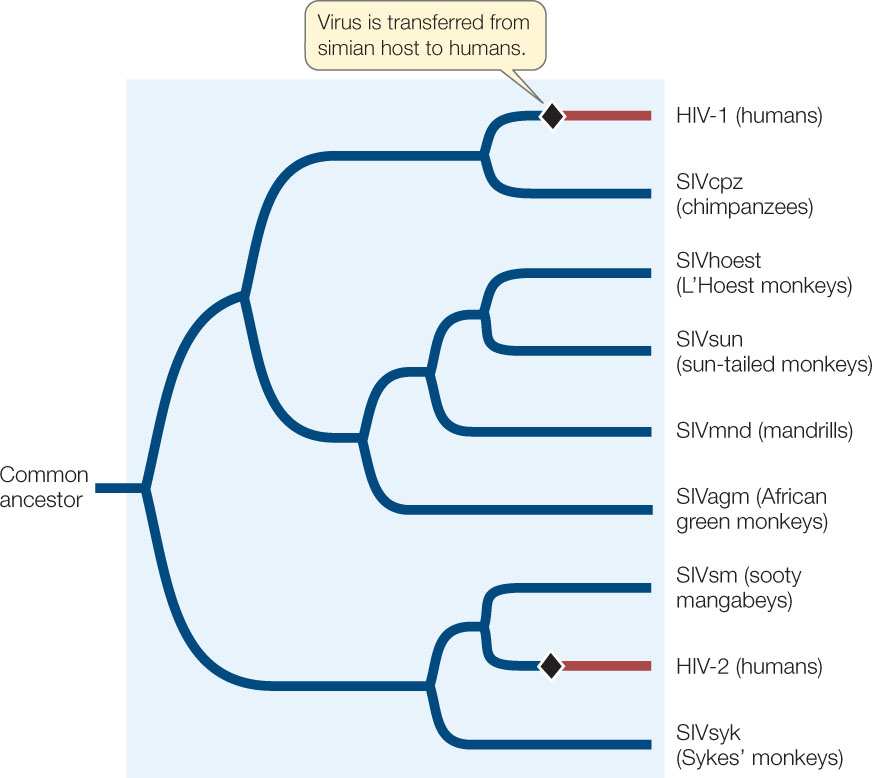
HIV-1 is the common form of the virus in human populations in central Africa, where chimpanzees are hunted for food, and HIV-2 is the common form in human populations in western Africa, where sooty mangabeys are hunted for food. Thus it seems likely that these viruses entered human populations through hunters who cut themselves while skinning chimpanzees and sooty mangabeys. The global pandemic of AIDS occurred when these infections in local African populations rapidly spread through human populations around the world.
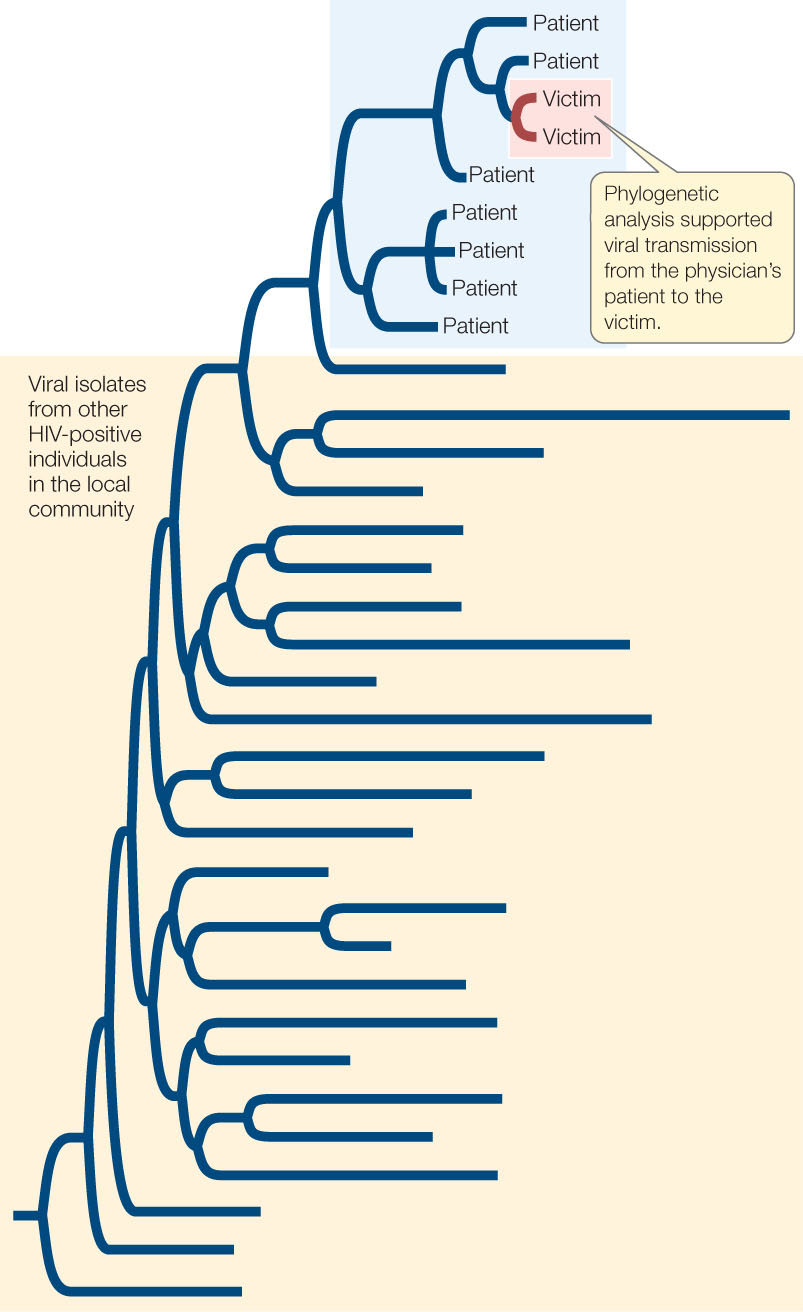
In recent years, phylogenetic analysis has become important in forensic investigations that involve viral transmission events. For example, phylogenetic analysis was critical for a criminal investigation of a physician who was accused of purposefully injecting blood from one of his HIV-positive patients into his former girlfriend in an attempt to kill her. The phylogenetic analysis revealed that the HIV strains present in the girlfriend were a subset of those present in the physician’s patient (FIGURE 16.7). Other evidence was needed, of course, to connect the physician to this purposeful transmission event, but the phylogenetic analysis was important to confirm the viral transmission event from the patient to the victim.
335
Phylogenies allow us to understand the evolution of complex traits
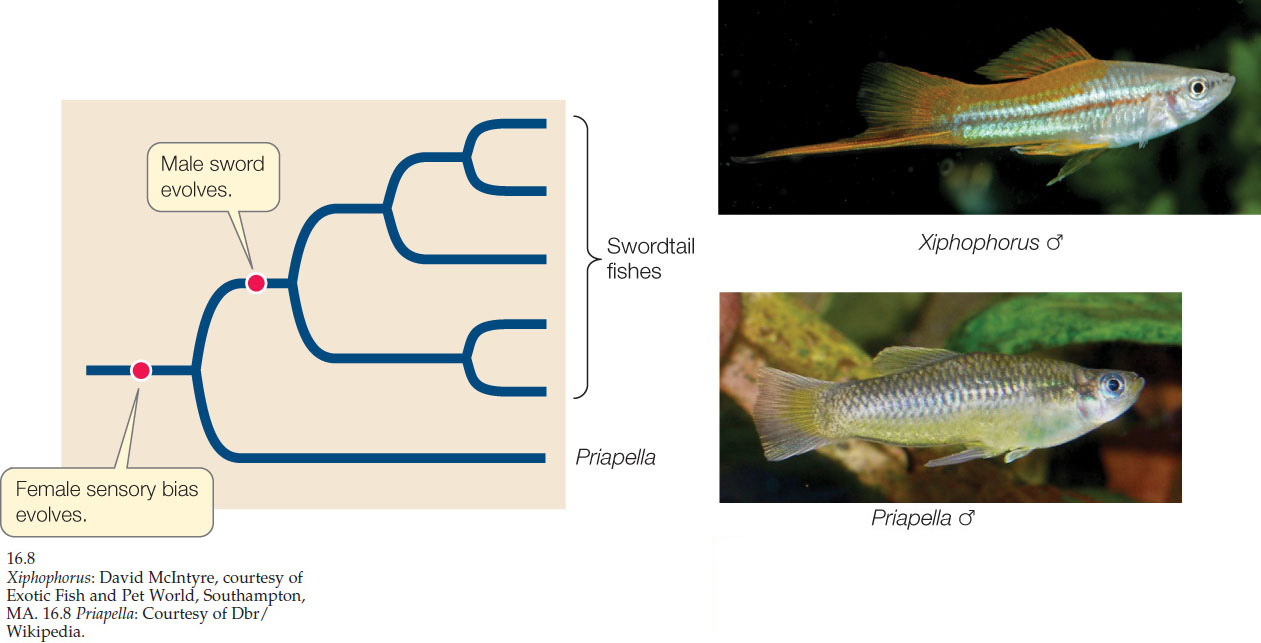
Male swordtails—a group of fishes in the genus Xiphophorus—have a long, colorful tail extension, and their reproductive success is closely associated with this appendage. Males with a long sword are more likely to mate successfully than are males with a short sword (an example of sexual selection; see Concept 15.2). Several explanations have been advanced for the evolution of this structure, including the hypothesis that the sword simply exploits a preexisting bias in the sensory system of the females. This sensory exploitation hypothesis suggests that female swordtails had a preference for males with long tails even before the tails evolved (perhaps because females assess the size of males by their total body length—including the tail—and prefer larger males).
To test the sensory exploitation hypothesis, phylogenetic analysis was used to identify the relatives of swordtails that had split most recently from their lineage before the evolution of swords. These closest relatives turned out to be fishes in the genus Priapella. Even though male Priapella do not normally have swords, when researchers attached artificial swordlike structures to the tails of male Priapella, female Priapella preferred those males. This result provided support for the hypothesis that female Xiphophorus had a preexisting sensory bias favoring tail extensions even before the trait evolved. Thus a long tail became a sexually selected trait because of the preexisting preference of the females (FIGURE 16.8).
336
Phylogenies can reveal convergent evolution
Like most animals, flowering plants (angiosperms) often reproduce by mating with another individual of the same species. But in many angiosperm species, the same individual produces both male and female gametes (contained within pollen and ovules, respectively). Self-incompatible species have mechanisms to prevent fertilization of the ovule by the individual’s own pollen, and so must reproduce by outcrossing with another individual. Individuals of some species, however, regularly fertilize their ovules using their own pollen; they are self-fertilizing or selfing species, and their gametes are self-compatible.
LINK
Some mechanisms of self-incompatibility are discussed in Concept 27.1
The evolution of angiosperm fertilization mechanisms was examined in Leptosiphon, a genus in the phlox family that exhibits a diversity of mating systems and pollination mechanisms. The self-incompatible (outcrossing) species of Leptosiphon have long petals and are pollinated by long-tongued flies. In contrast, self-pollinating species have short petals and do not require insect pollinators to reproduce successfully. Using nuclear ribosomal DNA sequences, investigators reconstructed the phylogeny of this genus (FIGURE 16.9). They then determined whether each species was self-compatible by artificially pollinating flowers with the plant’s own pollen or with pollen from other individuals and observing whether viable seeds formed.
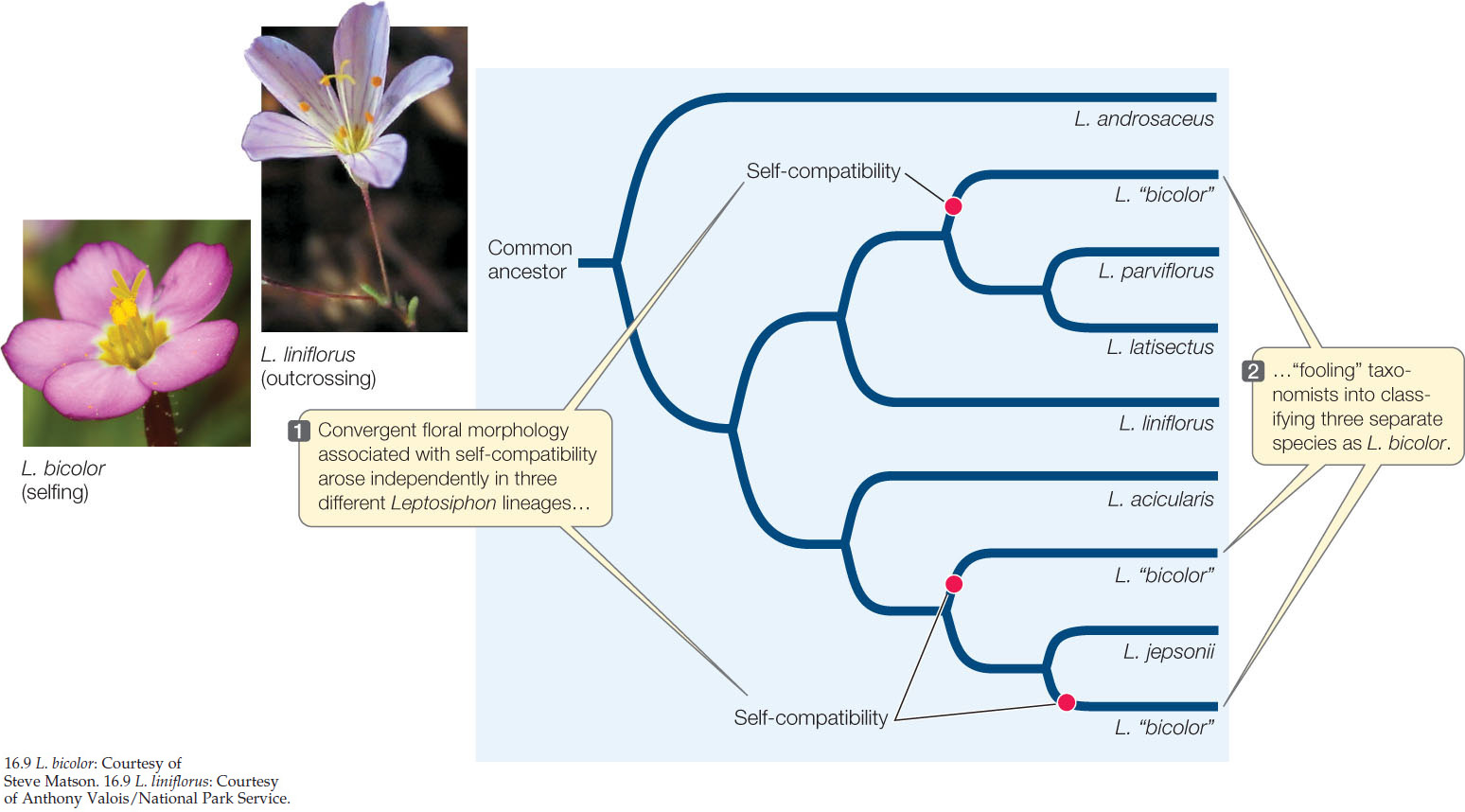
The reconstructed phylogeny suggests that self-incompatibility is the ancestral state and that self-compatibility evolved three times within this group of Leptosiphon. The change to self-compatibility eliminated the plants’ dependence on an outside pollinator and has been accompanied by the evolution of reduced petal size. Indeed, the striking morphological similarity of the flowers in the self-compatible groups once led to their being classified as members of a single species (L. bicolor). Phylogenetic analysis, however, shows them to be members of three distinct lineages. From this information we can infer that self-compatibility and its associated floral structure are convergent in the three independent lineages that had been called L. bicolor.
Ancestral states can be reconstructed
In addition to using phylogenetic methods to infer evolutionary relationships, biologists can use these techniques to reconstruct the morphology, behavior, or nucleotide and amino acid sequences of ancestral species (as was demonstrated for the ancestral sequence of bacteriophage T7 in Figure 16.5). In the opening of this chapter, we described how Mikhail Matz used phylogenetic analysis to reconstruct the sequence of changes in fluorescent proteins of corals to understand how red fluorescent proteins could be produced.
Reconstruction of ancient DNA sequences can also provide information about the biology of long-extinct organisms. For example, phylogenetic analysis was used to reconstruct an opsin protein in the ancestral archosaur (the most recent common ancestor of birds, dinosaurs, and crocodiles). Opsins are pigment proteins involved in vision; different opsins (with different amino acid sequences) are excited by different wavelengths of light. Knowledge of the opsin sequence in the ancestral archosaur would provide clues about the animal’s visual capabilities and therefore about some of its probable behaviors. Investigators used phylogenetic analysis of opsin from living vertebrates to estimate the amino acid sequence of the pigment that existed in the ancestral archosaur. A protein with this same sequence was then constructed in the laboratory. The investigators tested the reconstructed opsin and found a significant shift toward the red end of the spectrum in the light sensitivity of this protein compared with that of most modern opsins. Modern species that exhibit similar sensitivity are adapted for nocturnal vision, so the investigators inferred that the ancestral archosaur might have been active at night. Thus, reminiscent of the movie Jurassic Park, phylogenetic analyses are being used to reconstruct extinct species, one protein at a time.
337
Molecular clocks help date evolutionary events
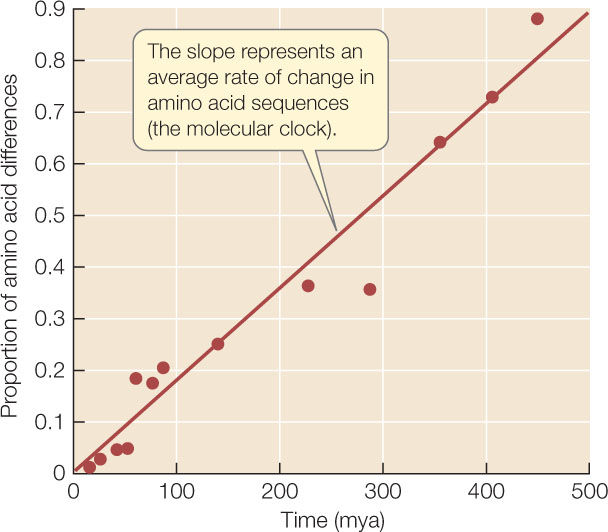
For many applications, biologists want to know not only the order in which evolutionary lineages split but also the timing of those splits. In 1965, Emile Zuckerkandl and Linus Pauling hypothesized that rates of molecular change were constant enough that they could be used to predict evolutionary divergence times—an idea that has become known as the molecular clock hypothesis.
Of course, different genes evolve at different rates, and there are also differences in evolutionary rates among species related to differing generation times, environments, efficiencies of DNA repair systems, and other biological factors. Nonetheless, among closely related species, a given gene usually evolves at a reasonably constant rate. Therefore the protein encoded by the gene accumulates amino acid replacements at a relatively constant rate (FIGURE 16.10). A molecular clock uses the average rate at which a given gene or protein accumulates changes to gauge the time of divergence for a particular split in the phylogeny. Molecular clocks must be calibrated using independent data, such as the fossil record, known times of divergence, or biogeographic dates (e.g., the time of separations of continents). Using such calibrations, times of divergence have been estimated for many groups of species that have diverged over millions of years.
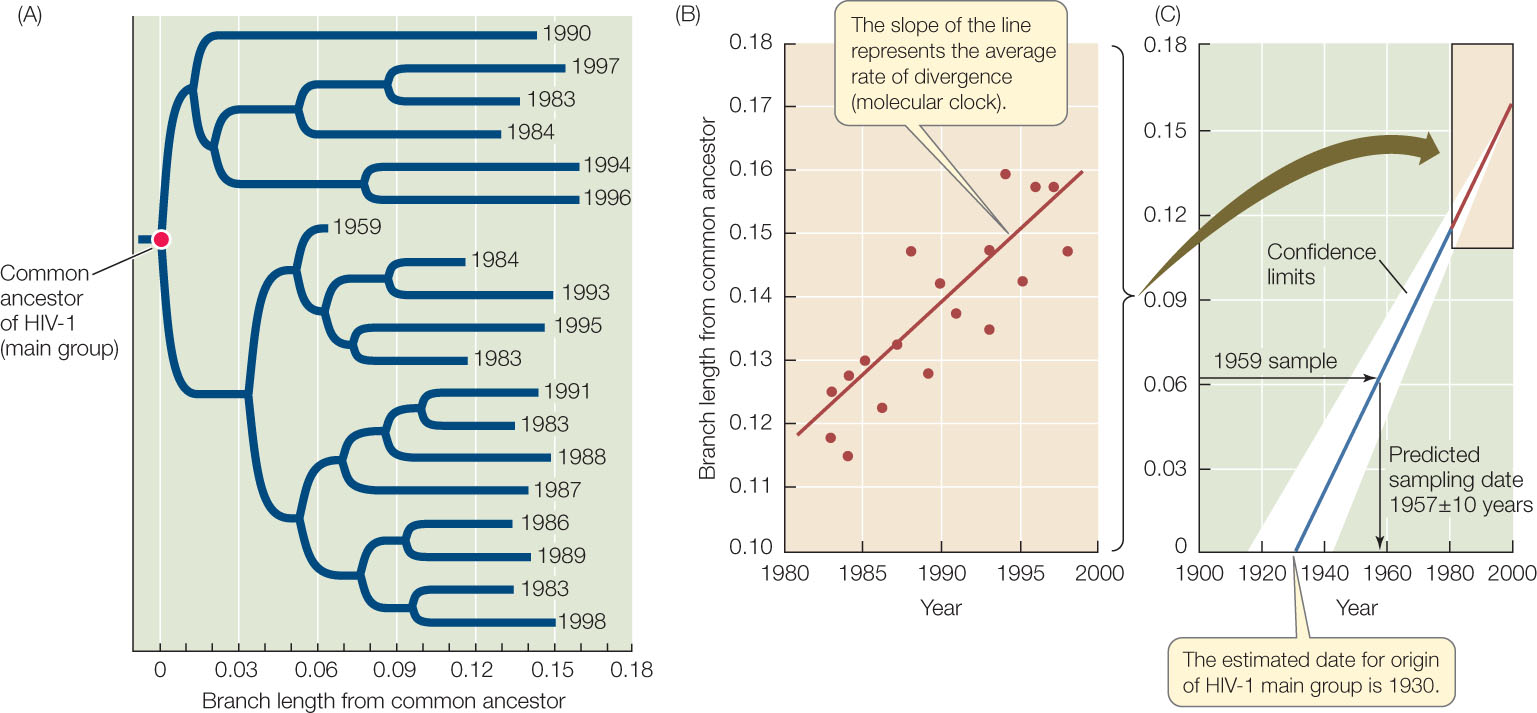
Molecular clocks are not only used to date ancient events; they are also used to study the timing of comparatively recent events. Most samples of HIV-1 have been collected from humans only since the early 1980s, although a few isolates from medical biopsies are available from as early as the 1950s. But biologists can use the observed changes in HIV-1 over the past several decades to project back to the common ancestor of all HIV-1 isolates, and estimate when HIV-1 first entered human populations from chimpanzees (FIGURE 16.11). This molecular clock was calibrated using the samples from the 1980s and 1990s, and then tested using the samples from the 1950s. As shown in Figure 16.11C, a sample from a 1959 biopsy is dated by molecular clock analysis at 1957 ± 10 years. Extrapolation back to the common ancestor of the samples suggested a date of origin for this group of viruses of about 1930. Although AIDS was unknown to Western medicine until the 1980s, this analysis shows that HIV-1 was present (probably at a very low frequency) in human populations in Africa for at least a half-century before its emergence as a global pandemic. Biologists have used similar analyses to conclude that immunodeficiency viruses have been transmitted repeatedly into human populations from multiple primates for more than a century (see also Figure 16.6).
338
CHECKpoint CONCEPT 16.3
- How can phylogenetic trees help determine the numberof times a particular trait evolved?
- How does the reconstruction of ancestral traits help biologists explain the biology of extinct species?
- What is the importance of adding a time dimension to phylogenetic trees, and how do biologists accomplish this?
All of life is connected through evolutionary history, and the relationships among organisms provide a natural basis for making biological comparisons. For these reasons, biologists use phylogenetic relationships as the basis for organizing life into a coherent classification system.