Chapter 1. Impact 8.2
Impact …ON BIOCHEMISTRY: I8.2 Proteins and nucleic acids
Polymers are macromolecules synthesized by stringing together and (in some cases) cross-linking smaller units known as monomers. There are macromolecules everywhere, inside us and outside us. Some are natural: they include cellulose, proteins, and deoxyribonucleic acid (DNA). Others are synthetic: they include polymers such as nylon and polystyrene. Here we describe the important role that hydrogen bonding plays in determining the shapes adopted by proteins and nucleic acids.
The concept of the ‘structure’ of a macromolecule takes on different meanings at the different levels at which we consider the arrangement of the chain or network of monomers. The term configuration refers to the structural features that can be changed only by breaking chemical bonds and forming new ones. Thus, the chains –A–B–C– and –A–C–B– have different configurations. The term conformation refers to the spatial arrangement of the different parts of a chain, and one conformation can be changed into another by rotating one part of a chain around a bond.
- The primary structure of a macromolecule is the sequence of small molecular residues making up the polymer.
For example, a protein is a polypeptide composed of linked α-amino acids, NH2CHRCOOH, where R is one of about 20 groups. The monomers are strung together by the peptide link,
–CONH–.
- The secondary structure of a macromolecule is the (often local) spatial arrangement of a chain.
The secondary structure of an isolated molecule of polyethylene is a random coil, whereas that of a protein is a highly organized arrangement determined largely by hydrogen bonds, and taking the form of random coils, helices (Fig. 1), or sheets in various segments of the molecule.
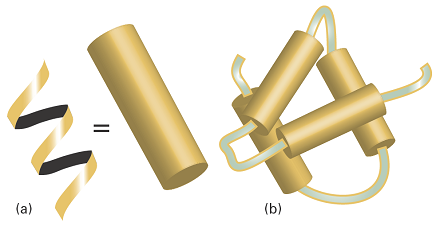
Fig. 1. (a) A polymer adopts a highly organized helical conformation, an example of a secondary structure. The helix is represented as a cylinder. (b) Several helical segments connected by short random coils pack together, providing an example of tertiary structure.
- The tertiary structure is the overall three-dimensional structure of a macromolecule.
For instance, the hypothetical protein shown in Fig. 1b has helical regions connected by short random-coil sections. The helices interact to form a compact tertiary structure.
- The quaternary structure of a macromolecule is the manner in which large molecules are formed by the aggregation of others.
Figure 2 shows how four molecular subunits, each with a specific tertiary structure, aggregate together. Quaternary structure can be very important in biology. For example, the oxygen-transport protein haemoglobin consists of four subunits that work together to take up and release O2.
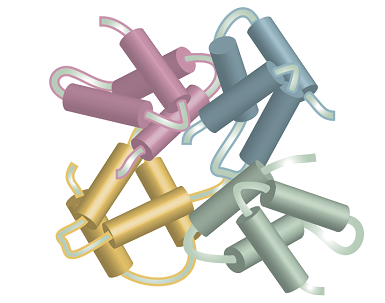
Fig. 2. Several subunits with specific tertiary structures pack together, providing an example of quaternary structure.
For a protein to function correctly, it needs to have a well-defined conformation. For example, an enzyme has its greatest catalytic efficiency only when it is in a specific conformation. The amino acid sequence of a protein contains the necessary information to create the active conformation of the protein from a newly synthesized random coil. However, the prediction of the conformation from the primary structure, the so-called protein folding problem, is extraordinarily difficult and is still the focus of much research.
The origin of the secondary structures of proteins is found in the rules formulated by Linus Pauling and Robert Corey in 1951. The essential feature is the stabilization of structures by hydrogen bonds involving the peptide link. The latter can act both as a donor of the H atom (the NH part of the link) and as an acceptor (the CO part). The Corey–Pauling rules are as follows (Fig. 3):
- The four atoms of the peptide link lie in a relatively rigid plane, which arises from delocalization of π electrons over the O, C, and N atoms and the maintenance of maximum overlap of their p orbitals.
- The N, H, and O atoms of a hydrogen bond lie in a straight line (with displacements of H tolerated up to not more than 30° from the N–O vector).
- All NH and CO groups are engaged in hydrogen bonding.
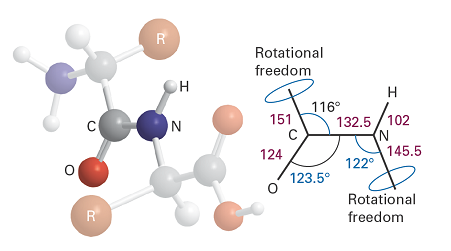
Fig. 3. The dimensions that characterise the peptide link. The C–NH–CO–C atoms define a plane (the C–N bond has partial double‑bond character), but there is rotational freedom around the C–CO and N–C bonds.
The rules are satisfied by two structures. One, in which hydrogen bonding between peptide links leads to a helical structure, is a helix, which can be arranged as either a right- or a left-handed screw. The other, in which hydrogen bonding between peptide links leads to a planar structure, is a sheet; this form is the secondary structure of the protein fibroin, the constituent of silk.
There is a barrier to internal rotation of one bond relative to another (just like the barrier to internal rotation in ethane). Because the planar peptide link is relatively rigid, the geometry of a polypeptide chain can be specified by the two angles that two neighbouring planar peptide links make to each other. Figure 4 shows the two angles \(\phi\) and ψ commonly used to specify this relative orientation. The sign convention is that a positive angle means that the front atom must be rotated clockwise to bring it into an eclipsed position relative to the rear atom. For an all-trans form of the chain, all \(\phi\) and ψ are 180°. A helix is obtained when all the \(\phi\) are equal and when all the ψ are equal. For a right-handed helix, all \(\phi\) = −57° and all ψ = −47°. For a left-handed helix, both angles are positive. The torsional contribution to the total potential energy of the molecule is
\(V_{torsion}= A(1 + cos 3 \phi ) + B(1+ cos 3 \psi )\)
in which A and B are constants of the order of 1 kJ mol−1. Because only two angles are needed to specify the conformation of a helix, and they range from −180° to +180° the torsional potential energy of the entire molecule can be represented on a Ramachandran plot, a contour diagram in which one axis represents \(\phi\) and the other represents ψ.
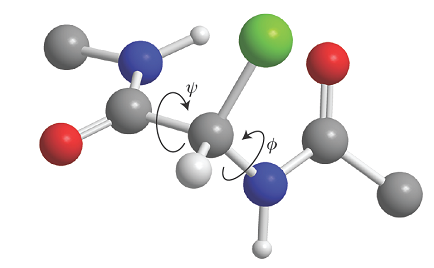
Fig. 4. The definition of the torsional angles ψ and \(\phi\) between two peptide units. In this case (an α‑L‑polypeptide) the chain has been drawn in its all‑trans form, with ψ = \(\phi\) = 180°.
A right-handed α-helix is illustrated in Fig. 5. Each turn of the helix contains 3.6 amino acid residues, so the period of the helix corresponds to 5 turns (18 residues). The pitch of a single turn (the distance between points separated by 360°) is 544 pm. The N–H…O bonds lie parallel to the axis and link every fourth group (so residue i is linked to residues i − 4 and i + 4). All the R groups point away from the major axis of the helix.
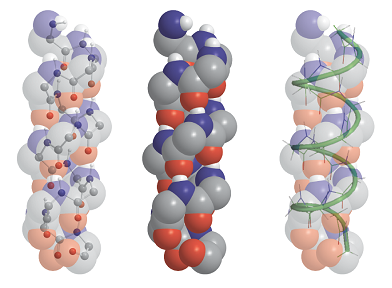
Fig. 5. The polypeptide α helix, with poly-L-glycine as an example. There are 3.6 residues per turn, and a translation along the helix of 150 pm per residue, giving a pitch of 540 pm. The diameter (ignoring side chains) is about 600 pm.
Figure 6 shows the Ramachandran plots for the helical form of polypeptide chains formed from the nonchiral amino acid glycine (R = H) and the chiral amino acid L-alanine (R = CH3). The glycine map is symmetrical, with minima of equal depth at \(\phi\) = −80°, ψ = +90° and at \(\phi\) = +80°, ψ = −90°. In contrast, the map for L-alanine is unsymmetrical, and there are three distinct low-energy conformations (marked I, II, III). The minima of regions I and II lie close to the angles typical of right- and left-handed helices, but the former has a lower minimum. This result is consistent with the observation that polypeptides of the naturally occurring L-amino acids tend to form right-handed helices.
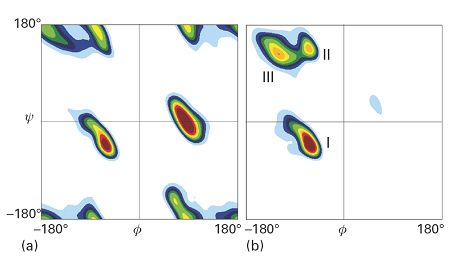
Fig. 6. Contour plots of potential energy against the angles ψ and \(\phi\), also known as a Ramachandran diagram, for (a) a glycyl residue of a polypeptide chain and (b) an alanyl residue. The glycyl diagram is symmetrical, but that for alanyl is unsymmetrical and the global minimum corresponds to an α-helix. (T. Hovmoller et al., Acta Cryst. D58, 768 (2002).)
A β-sheet (also called the β-pleated sheet) is formed by hydrogen bonding between two extended polypeptide chains (large absolute values of the torsion angles \(\phi\) and ψ). Some of the R groups point above and some point below the sheet. Two types of structures can be distinguished from the pattern of hydrogen bonding between the constituent chains.
In an anti-parallel β-sheet (Fig. 7a), \(\phi\) = −139o, ψ = 113o, and the N–H…O atoms of the hydrogen bonds form a straight line. This arrangement is a consequence of the antiparallel arrangement of the chains: every N–H bond on one chain is aligned with a C–O bond from another chain. Antiparallel β-sheets are very common in proteins. In a parallel β-sheet (Fig. 7b), \(\phi\) = −119o, ψ = 113o, and the N–H…O atoms of the hydrogen bonds are not perfectly aligned. This arrangement is a result of the parallel arrangement of the chains: each N–H bond on one chain is aligned with an N–H bond of another chain and, as a result, each C–O bond of one chain is aligned with a C–O bond of another chain. These structures are not common in proteins.
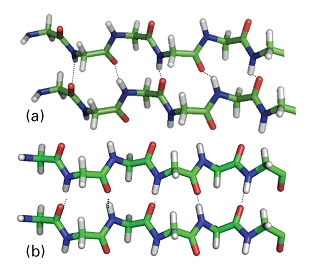
Fig. 7. The two types of β-sheets: (a) antiparallel (\(\phi\) = –139°, ψ = 113°), in which the N–H–O atoms of the hydrogen bonds form a straight-line; (b) parallel (\(\phi\) = –119°, ψ = 113°) in which the N–H–O atoms of the hydrogen bonds are not perfectly aligned.
Although all the rules that govern protein folding are not known, a few general conclusions may be drawn from X-ray diffraction studies of water-soluble natural proteins and synthetic polypeptides. In an aqueous environment, the chains fold in such a way as to place nonpolar R groups in the interior (which is often not very accessible to solvent) and charged R groups on the surface (in direct contact with the polar solvent). Other factors that promote the folding of proteins include covalent disulfide (–S–S–) links, Coulombic interactions between ions (which depend on the degree of protonation of groups and therefore on the pH), hydrogen bonding, van der Waals interactions, and solvent effects.
Nucleic acids are key components of the mechanism of storage and transfer of genetic information in biological cells. Deoxyribonucleic acid contains the instructions for protein synthesis, which is carried out by different forms of ribonucleic acid (RNA). In this section, we discuss the main structural features of DNA and RNA.
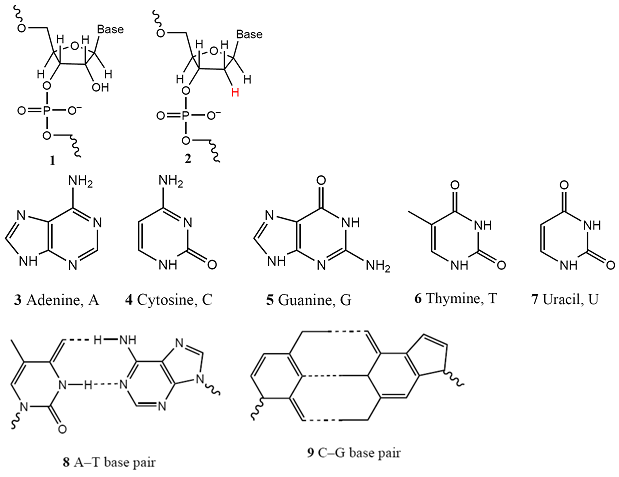
Both DNA and RNA are polynucleotides (1), in which base-sugar-phosphate units are linked by phosphodiester bonds. In RNA the sugar is β-D-ribose and in DNA it is β-D-2-deoxyribose (as shown in 2). The most common bases are adenine (A, 3), cytosine (C, 4), guanine (G, 5), thymine (T, found in DNA only, 6), and uracil (U, found in RNA only, 7). At physiological pH, each phosphate group of the chain carries a negative charge and the bases are deprotonated and neutral. This charge distribution leads to two important properties. One is that the polynucleotide chain is a polyelectrolyte, a macromolecule with many different charged sites, with a large and negative overall surface charge. The second is that the bases can interact by hydrogen bonding, as shown for A–T (8) and C–G base pairs (9). The secondary and tertiary structures of DNA and RNA arise primarily from the pattern of this hydrogen bonding between bases of one or more chains.
In DNA, two polynucleotide chains wind around each other to form a double helix (Fig. 8). The chains are held together by links involving A–T and C–G base pairs that lie parallel to each other and perpendicular to the major axis of the helix. The structure is stabilized further by base-stacking, in which dispersion interactions bring together the planar π systems of bases. In B-DNA, the most common form of DNA found in biological cells, the helix is right-handed with a diameter of 2.0 nm and a pitch of 3.4 nm. Long stretches of DNA can fold further into a variety of tertiary structures. Supercoiled DNAis found in the chromosome and can be visualized as the twisting of closed circular DNA (ccDNA), much like the twisting of a rubber band.
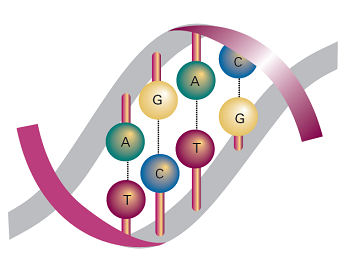
Fig. 8 DNA double helix, in which two polynucleotide chains are linked together by hydrogen bonds between adenine (A) and thymine (T) and between cytosine (C) and guanine (G).
The extra –OH group in β-D-ribose imparts enough steric strain to a polynucleotide chain that stable double helices cannot form in RNA. Therefore, RNA exists primarily as single chains that can fold into complex structures by formation of A–U and G–C base pairs. One example of this is the structure of transfer RNA (tRNA), in which base-paired regions are connected by loops and coils. Transfer RNAs help assemble polypeptide chains during protein synthesis in the cell.