35.2 DNA Damage Can Be Detected and Repaired
✓ 5 Explain the mechanisms that ensure the fidelity of DNA replication.
To protect the integrity of the genetic message, a wide range of DNA-repair systems are present in most organisms. The bacterium Deinococcus radiodurans illustrates the extraordinary power of such repair systems. This bacterium was discovered in 1956 when scientists were studying the use of high doses of gamma radiation to sterilize canned meat. In some cases, the meat still spoiled due to the growth of a bacterial species that withstood doses of gamma radiation more than 1000 times larger than those that would kill a human being. Even when the bacterial chromosomes are broken into many fragments by the ionizing radiation, they can reassemble and recombine to regenerate the intact genome with essentially no loss of information. While most organisms do not have the robust repair systems of D. radiodurans, all organisms require the ability to repair damaged DNA.
Many systems repair DNA by using sequence information from the uncompromised strand. Such single-strand replication systems follow a similar mechanistic outline:
Recognize the offending base(s).
Remove the offending base(s).
Repair the resulting gap with a DNA polymerase and DNA ligase.
We will briefly consider examples of several repair pathways that follow these basic steps. Although many of these examples are taken from E. coli, corresponding repair systems are present in most other organisms, including human beings.
The replicative DNA polymerases are able to correct many DNA mismatches produced in the course of replication. For example, a subunit of E. coli DNA polymerase III functions as a 3′-to-5′exonuclease, as described in Chapter 34. This domain removes mismatched nucleotides from the 3′ end of DNA by hydrolysis. How does the enzyme sense whether a newly added base is correct? As a new strand of DNA is synthesized, it is proofread. If an incorrect base is inserted, then DNA synthesis slows down and the mismatched pair, which is weakly bound, is able to fluctuate in position. The delay from the slowdown allows time for these fluctuations to take the newly synthesized strand out of the polymerase active site and into the exonuclease active site (Figure 34.9). There, the DNA is degraded until it moves back into the polymerase active site and synthesis continues.
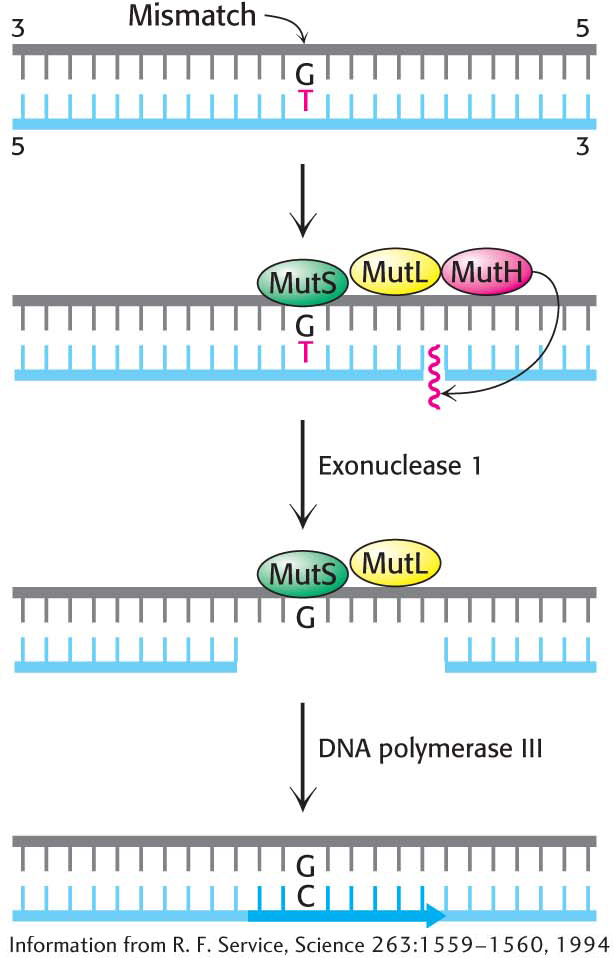
Figure 35.7: Mismatch repair. DNA mismatch repair in E. coli is initiated by the interplay of MutS, MutL, and MutH proteins. A G–T mismatch is recognized by MutS. MutL binds to MutS and activates the MutH nuclease. MutH cleaves the backbone in the vicinity of the mismatch. A segment of the DNA strand containing the erroneous T is removed by exonuclease I and synthesized anew by DNA polymerase III.
A second mechanism is present in essentially all cells to correct errors made in replication that are not corrected by proofreading (Figure 35.7). Mismatch-repair systems consist of at least two proteins, one for detecting the mismatch and the other for recruiting an endonuclease that cleaves the newly synthesized DNA strand close to the lesion. An exonuclease can then excise the incorrect base, and a polymerase fills the gap. In E. coli, the mismatch-repair proteins are called MutS and MutL and the endonuclease is called MutH.
How does the mismatch-repair machinery determine which of the bases is incorrect? The answer is not well established for higher organisms; however, in E. coli, some adenine bases of the parent strand of DNA are methylated, whereas the newly synthesized daughter strand is not yet methylated. Thus, the repair machinery recognizes that the base attached to the methylated DNA is correct and removes the offending base from the daughter strand.
Sometimes, the damage to DNA can be repaired directly, without having to remove any fragments of the DNA, a process called direct repair. An example of direct repair is the photochemical cleavage of pyrimidine dimers by a photoreactivating enzyme called DNA photolyase. The E. coli enzyme, a 35-kDa protein that contains bound N5, N10-methenyltetrahydrofolate and flavin adenine dinucleotide (FAD) cofactors, binds to the distorted region of DNA. The enzyme uses light energy—specifically, the absorption of a photon by the N5, N10-methenyltetrahydrofolate coenzyme—to form an excited state that cleaves the dimer into its component bases.
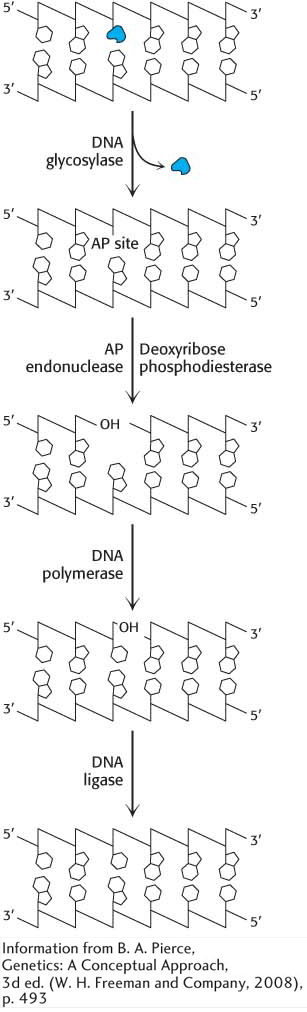
Figure 35.8: Base-excision repair. The base-excision-repair process identifies and removes a modified base. Subsequently, the entire nucleotide is replaced.
Modified bases, such as 8-oxyguanine or 3-methyladenine, are excised by the E. coli enzyme AlkA, an example of base-excision repair. The binding of this enzyme to damaged DNA flips the affected base out of the DNA double helix and into the active site of the enzyme. The enzyme then acts as a glycosylase, cleaving the glycosidic bond to release the damaged base. At this stage, the DNA backbone is intact, but a base is missing. This hole is called an AP site because it is apurinic (devoid of A or G) or apyrimidinic (devoid of C or T). An AP endonuclease recognizes this defect and nicks the backbone adjacent to the missing base. Deoxyribose phosphodiesterase excises the residual deoxyribose phosphate unit, and DNA polymerase I inserts an undamaged nucleotide, as dictated by the base on the undamaged complementary strand. Finally, the repaired strand is sealed by DNA ligase (Figure 35.8).
Base-excision repair corrects the most common point mutation in humans, C → T. Cytosine in eukaryotic DNA is frequently methylated in the 5 position as a means of transcription regulation. When 5-methylcytosine spontaneously deaminates, thymine is formed, generating a T–G base pair (Figure 35.9). Inasmuch as both T and G are normal bases in DNA, how does the repair machinery know which base to retain and which to remove? Because the C → T mutation is so common, the T in a T–G pair is always treated as the incorrect base and removed by the base-excision-repair proteins.
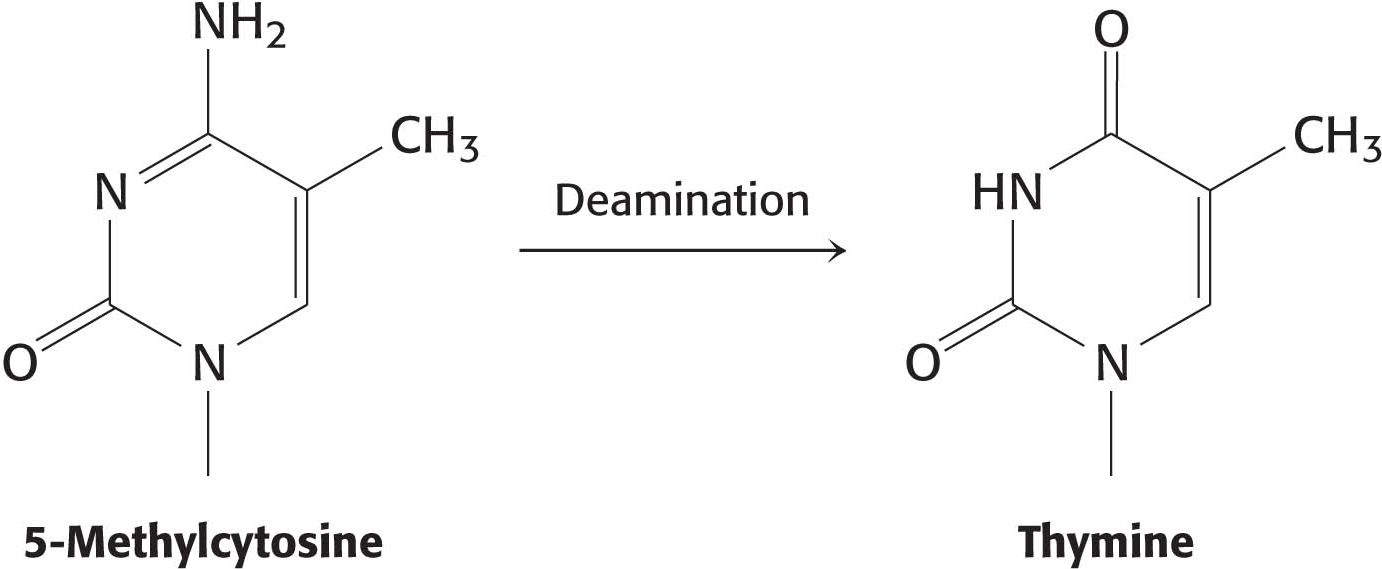
Figure 35.9: The deamination of 5-methylcytosine forms thymine.
Base-excision repair requires recognition of an altered base. However, not all damaged bases are recognized by DNA glycosylase. What system recognizes improper nucleotide pairs that escape the base-excision-repair system? The nucleotide-excision-repair system recognizes distortions in the DNA double helix caused by the presence of a damaged base. One of the best-understood examples of nucleotide-excision repair is utilized for the excision of a pyrimidine dimer. Three enzymatic activities are essential for this repair process in E. coli (Figure 35.10). First, an enzyme complex consisting of the proteins encoded by the uvrABC genes detects the distortion produced by the DNA damage. The UvrABC enzyme, an excinuclease (from the Latin exci, meaning “to cut out”), cuts the damaged DNA strand at two sites, eight nucleotides away from the damaged site on the 5′ side and four nucleotides away on the 3′ side. DNA polymerase I then enters the gap to carry out repair synthesis. The 3′ end of the nicked strand is the primer, and the intact complementary strand is the template. Finally, the 3′ end of the newly synthesized stretch of DNA and the original part of the DNA chain are joined by DNA ligase.
!quickquiz! QUICK QUIZ 1
What property of the structure of DNA allows for the repair of bases damaged by mutation?
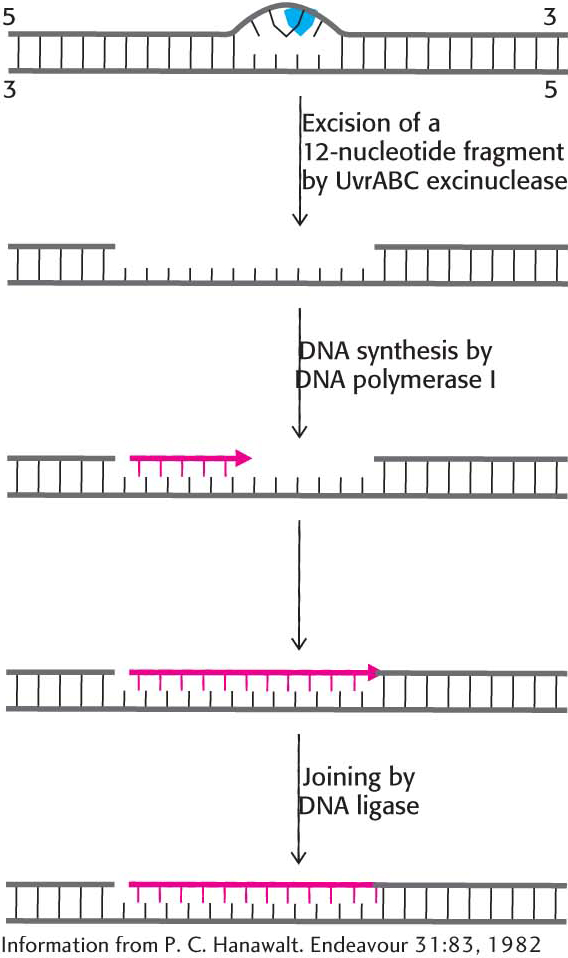
Figure 35.10: Nucleotide-excision repair. The repair of a region of DNA containing a thymine dimer by the sequential action of a specific excinuclease, a DNA polymerase, and a DNA ligase. The thymine dimer is shown in blue, and the new region of DNA is in red.
The Presence of Thymine Instead of Uracil in DNA Permits the Repair of Deaminated Cytosine
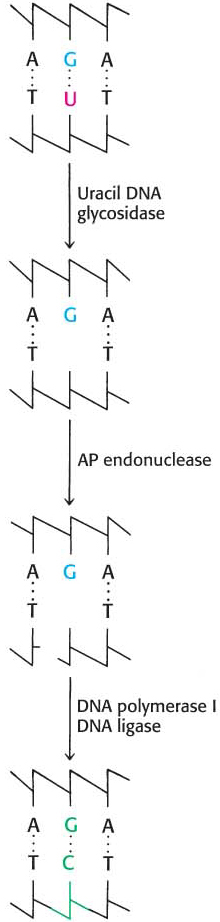
Figure 35.11: Uracil repair. Uridine bases in DNA, formed by the deamination of cytidine, are excised and replaced by cytidine.
The presence in DNA of thymine rather than uracil was an enigma for many years. Both bases pair with adenine. The only difference between them is a methyl group in thymine in place of the C-5 hydrogen atom in uracil. Why is a methylated base employed in DNA and not in RNA? The existence of an active repair system to correct the deamination of cytosine provides a convincing solution to this puzzle.
Cytosine in DNA spontaneously deaminates at a perceptible rate to form uracil. The deamination of cytosine is potentially mutagenic because uracil pairs with adenine, and so one of the daughter strands will contain a U–A base pair rather than the original C–G base pair. This mutation is prevented by a base-excision-repair system that recognizes uracil to be foreign to DNA (Figure 35.11). The repair enzyme, uracil DNA glycosylase, hydrolyzes the glycosidic bond between the uracil and deoxyribose moieties but does not attack thymine-containing nucleotides. The AP site generated is repaired to reinsert cytosine. Thus, the methyl group on thymine is a tag that distinguishes thymine from deaminated cytosine. If thymine were not used in DNA, uracil correctly in place would be indistinguishable from uracil formed by deamination. The defect would persist unnoticed, and so a C–G base pair would necessarily be mutated to U–A in one of the daughter DNA molecules. Thymine is used instead of uracil in DNA to preserve the fidelity of the genetic message.
!clinic! CLINICAL INSIGHT: Many Cancers Are Caused by the Defective Repair of DNA
As described in Chapter 13, cancers are caused by mutations in genes associated with growth control. Defects in DNA-repair systems increase the overall frequency of mutations and, hence, the likelihood of cancer-causing mutations. Indeed, the synergy between studies of mutations that predispose people to cancer and studies of DNA repair in model organisms have had a tremendous impact in revealing the biochemistry of DNA-repair pathways. Genes for DNA-repair proteins are often tumor-suppressor genes; that is, they suppress tumor development when at least one copy of the gene is free of a deleterious mutation. When both copies of a gene are mutated, however, tumors develop at rates greater than those for the population at large. People who inherit defects in a single tumor-suppressor allele do not necessarily develop cancer but are susceptible to developing the disease because only the one remaining normal copy of the gene must mutate to further the development of cancer.
Consider, for example, xeroderma pigmentosum, a rare human skin disease (Figure 35.12). The skin in an affected person is extremely sensitive to sunlight or ultraviolet light. In infancy, severe changes in the skin become evident and worsen with time. The skin becomes dry, and there is a marked atrophy of the dermis. Keratoses (skin growth derived from epidermal keratinocytes) appear, the eyelids become scarred, and the cornea ulcerates. Skin cancer usually develops at several sites. Many patients die before age 30 from metastases of these malignant skin tumors. Studies of xeroderma pigmentosum patients have revealed that mutations occur in genes for a number of different proteins. These proteins are components of the human nucleotide-excision-repair pathway, including components similar to the UvrABC subunits.
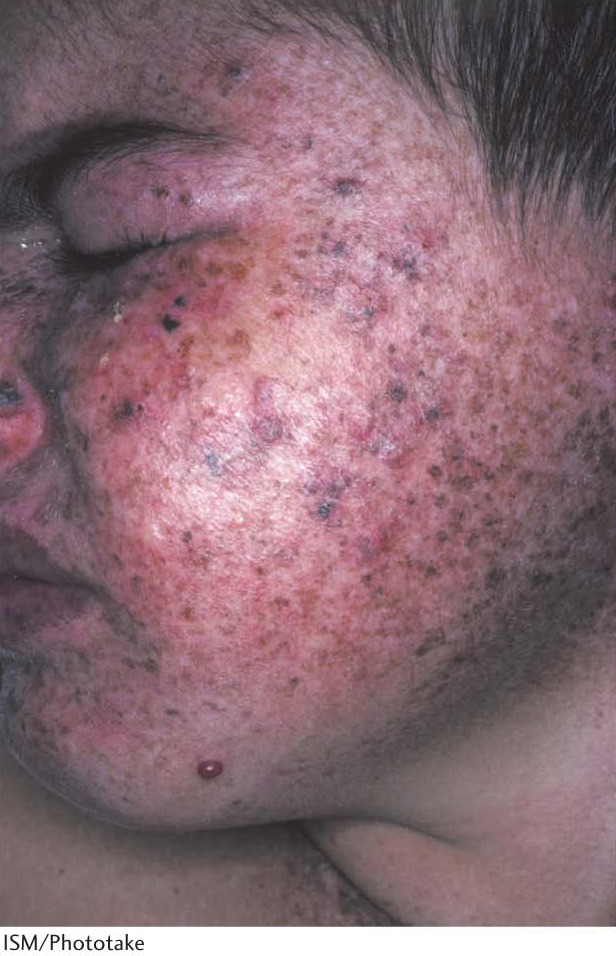
Figure 35.12: Xeroderma pigmentosa. In people suffering from xeroderma pigmentosa, numerous hyperpigmented blemishes appear most commonly on sun-exposed skin. These blemishes may progress to rough-surfaced growths (solar keratoses) and, finally, to skin malignancies.
Defects in other repair systems can increase the frequency of other tumors. For example, hereditary nonpolyposis colorectal cancer (HNPCC, or Lynch syndrome) results from defective DNA mismatch repair. HNPCC is not rare—as many as 1 in 200 people will develop this form of cancer. Mutations in two genes, called hMSH2 and hMLH1, account for most cases of this hereditary predisposition to cancer. These mutated genes seem likely to allow mutations to accumulate throughout the genome. In time, genes important in controlling cell proliferation become altered, resulting in the onset of cancer.
The absence of repair mechanisms and the high rates of cell duplication in cancer cells present a therapeutic opportunity. Several agents widely used in cancer chemotherapy, including cyclophosphamide and cisplatin, act by damaging DNA. Ideally, the cancer cells are irreparably damaged and die.
!clinic! CLINICAL INSIGHT: Many Potential Carcinogens Can Be Detected by Their Mutagenic Action on Bacteria
Many human cancers are caused by exposure to chemicals that cause mutations. It is important to identify compounds that can cause mutations and ascertain their potency so that human exposure to them can be minimized. Bruce Ames devised a simple and sensitive test for detecting chemical mutagens. In the Ames test, a thin layer of agar containing about 109 bacteria of a specially constructed strain of Salmonella is placed on a Petri plate. These bacteria are unable to grow in the absence of histidine because a mutation is present in one of the genes for the biosynthesis of this amino acid. The addition of a chemical mutagen to the plate results in many new mutations. A small proportion of them reverse the original mutation, and histidine can be synthesized. These revertants multiply in the absence of an external source of histidine and appear as discrete colonies after the plate has been incubated at 37°C for 2 days (Figure 35.13). For example, a sample of 2-aminoanthracene, a known cancer-causing agent (carcinogen), gives 11,000 revertant colonies, compared with only 30 spontaneous revertants in its absence. A series of concentrations of a chemical can be readily tested to generate a dose–response curve. These curves are usually linear, which suggests that there is no threshold concentration for mutagenesis.
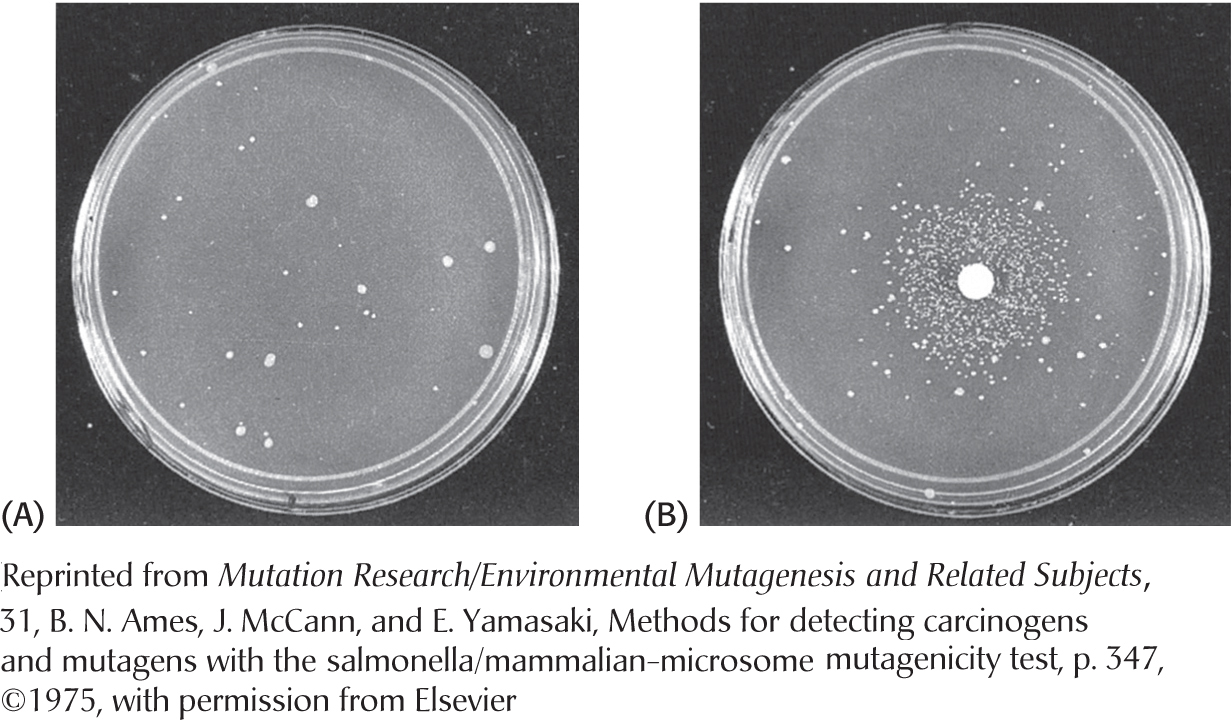
Figure 35.13: Ames test. (A) A petri plate containing about 109 Salmonella bacteria that cannot synthesize histidine. (B) The same petri plate with the addition of a filter-paper disc with a mutagen, which produces a large number of revertants that can synthesize histidine. After 2 days, the revertants appear as rings of colonies around the disc. The small number of visible colonies in plate A are spontaneous revertants.
A key feature of this detection system is the inclusion of a mammalian liver homogenate. Recall that some potential carcinogens such as aflatoxin are converted into their active forms by P450 enzyme systems in the liver or other mammalian tissues. Bacteria lack these enzymes, and so the test plate requires a few milligrams of a liver homogenate to activate this group of mutagens.
The Salmonella test is extensively used to help evaluate the mutagenic and carcinogenic risks of a large number of chemicals. This rapid and inexpensive bacterial assay for mutagenicity complements epidemiological surveys and animal tests that are necessarily slower, more laborious, and far more expensive.