Temperature Enhances the Rate of Enzyme-Catalyzed Reactions
As the temperature rises, the rate of most reactions increases. The rise in temperature increases the Brownian motion of the molecules, which makes interactions between an enzyme and its substrate more likely. For most enzymes, there is a temperature at which the increase in catalytic activity ceases and there is a precipitous loss of activity (Figure 8.1). What is the basis of this loss of activity? Recall from Chapter 4 that proteins have a complex three-dimensional structure that is held together by weak bonds. When the temperature increases beyond a certain point, the bonds maintaining the three-dimensional structure are not strong enough to withstand the polypeptide chain’s thermal jostling and the protein loses the structure required for activity. The protein is said to be denatured.
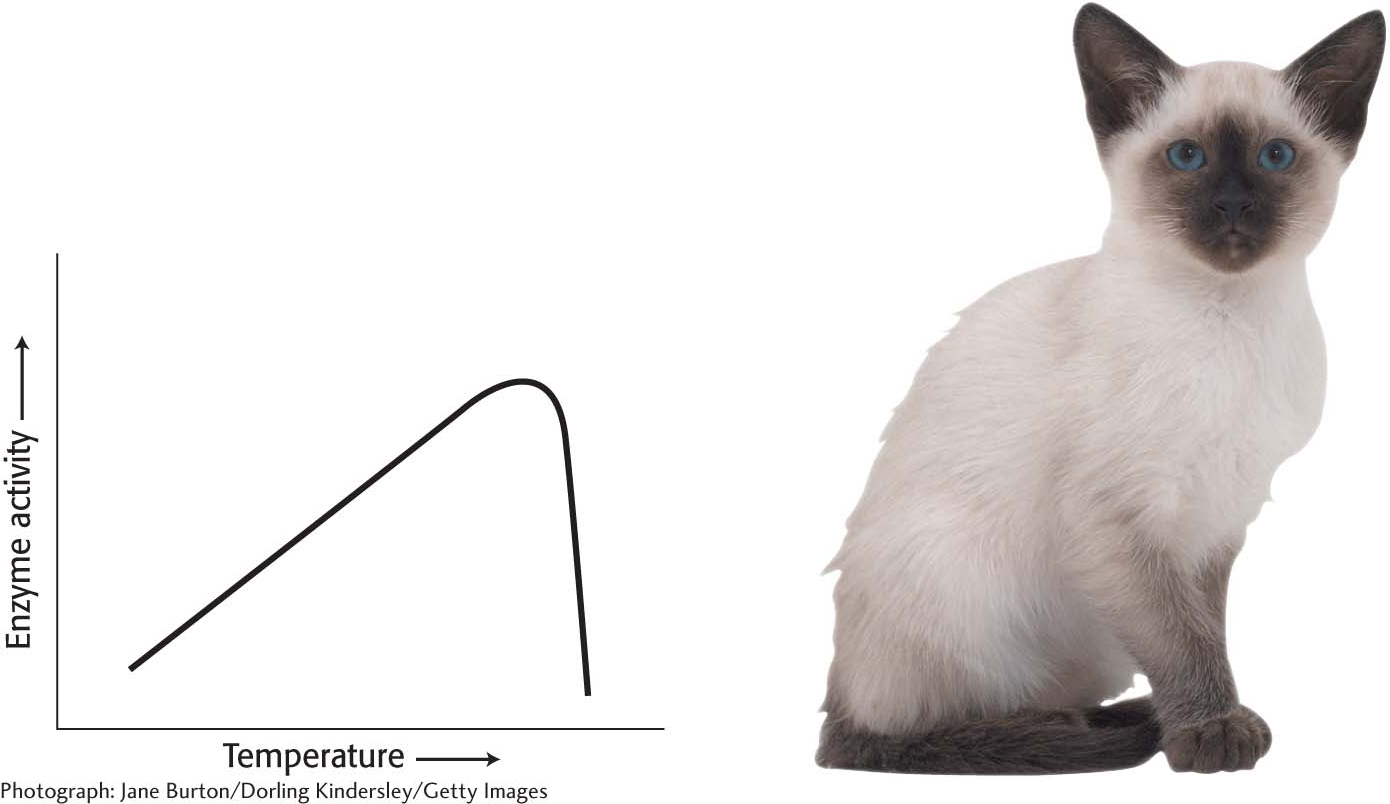
Figure 8.1: The effect of heat on enzyme activity. Enzyme activity increases with temperature until the enzyme is denatured. The enzyme tyrosinase, which is part of the pathway that synthesizes the pigment that results in dark fur, has a low tolerance for heat in Siamese cats. It is inactive at normal body temperatures but functional at slightly lower temperatures. The extremities of a Siamese cat are cool enough for tyrosinase to gain function and produce pigment.

Figure 8.2: A lizard basking in the sun. Ectotherms, such as the Namibian rock agama (Agama planiceps), adjust their body temperatures and, hence, the rate of biochemical reactions behaviorally.
In organisms such as ourselves that maintain a constant body temperature (endotherms), the effect of outside temperature on enzyme activity is minimized. However, in organisms that assume the temperature of the ambient environment (ectotherms), temperature is an important regulator of biochemical and, indeed, biological activity. Lizards, for instance, are most active in warmer temperatures and relatively inactive in cooler temperatures, a behavioral manifestation of biochemical activity (Figure 8.2). Although endotherms are not as sensitive to ambient temperature as ectotherms, slight tissue temperature alterations are sometimes important. For instance, when athletes “warm-up,” they are increasing the temperature of their muscles through exertion. This increase in temperature facilitates the biochemistry that will power the exercise.
Some organisms, such as thermophilic archaea, can live at temperatures of 80°C or higher, temperatures that would denature most proteins. The proteins in these organisms have evolved to be very resistant to thermal denaturation (Figure 8.3).

Figure 8.3: Thermophilic archaea and their environment. Archaea can thrive in habitats as harsh as a volcanic vent. Here, the archaea form an orange mat surrounded by yellow sulfurous deposits.
Most Enzymes Have an Optimal pH
Enzyme activity also often varies with pH, the H+ concentration of the environment. The activity of most enzymes displays a bell-shaped curve when examined as a function of pH. However, the optimal pH—the pH at which enzymes display maximal activity—varies with the enzyme and is correlated with the environment of the enzyme. For instance, the protein-digesting enzyme pepsin functions in the highly acidic environment of the stomach, where the pH is between 1 and 2. Most proteins are denatured at this pH, but pepsin functions very effectively. The enzymes of the pancreatic secretion of the upper small intestine, such as chymotrypsin, have pH optima near pH 8, in keeping with the pH of the intestine in this region (Figure 8.4).
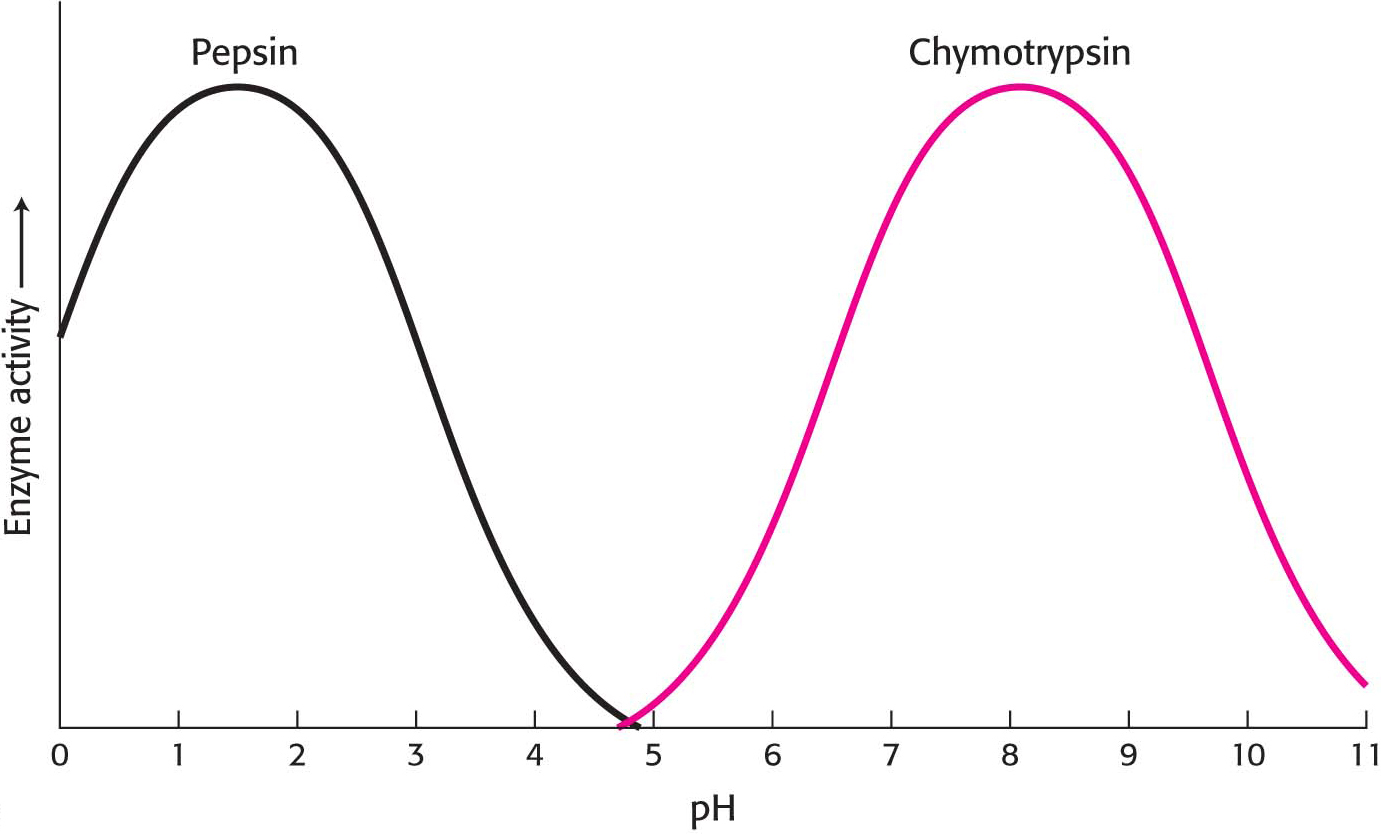
Figure 8.4: The pH dependence of the activity of the enzymes pepsin and chymotrypsin. Chymotrypsin and pepsin have different optimal pH values. The optimal pH for pepsin is noteworthy. Most proteins would be denatured at this acidic pH.
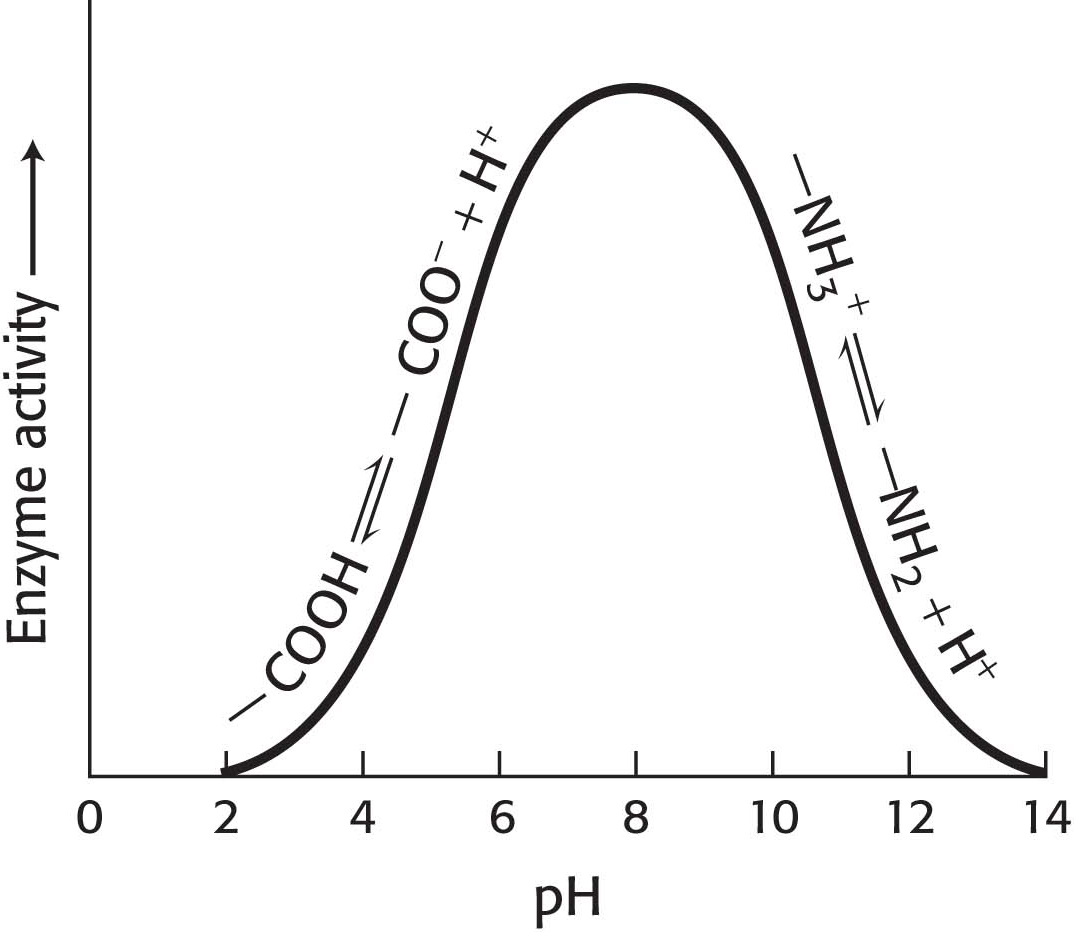
Figure 8.5: A pH profile for a hypothetical enzyme. The pH dependence of enzymes is due to the presence of ionizable R groups.
How can we account for the pH effect on enzyme activity in regard to our understanding of enzymes in particular and proteins in general? Imagine an enzyme that requires the ionization of both a glutamic acid residue and a lysine residue at the active site for the enzyme to be functional. Thus, the enzyme would depend on the presence of a—COO– group as well as an—NH3+ group (Figure 8.5). If the pH is lowered (the H+ concentration increases), the—COO− groups will gradually be converted into—COOH groups with a concomitant loss of enzyme activity. On the other side of the optimum, as the pH is raised (less H+, more OH−), the—NH3+ group loses an H+ to OH−, becoming a neutral—NH2 group, and the enzyme activity is diminished. Often, the activity-versus-pH curves are due to several ionizable groups.
Is alteration of the pH of the cellular milieu ever used as a regulator device? The answer is yes, and a crucial enzyme in glucose metabolism in skeletal muscle provides an example. Phosphofructokinase (Chapter 16) controls the rate of metabolism of glucose under aerobic conditions (in the presence of oxygen) and under anaerobic conditions (in the absence of oxygen). A problem arises with the rapid processing of glucose in the absence of oxygen: the end product is lactic acid, which readily ionizes to lactate and a hydrogen ion. To prevent the muscle from becoming “pickled” by the high concentration of acid, the activity of phosphofructokinase decreases if the pH falls too drastically, which, in turn, reduces the metabolism of glucose to lactic acid. Phosphofructokinase is made up of multiple subunits, and the decrease in pH causes the subunits to dissociate, rendering the enzyme inactive and thus reducing lactic acid production.
Enzymes Can Be Inhibited by Specific Molecules
The activity of many enzymes can be inhibited by the binding of specific small molecules and ions. This means of enzyme inhibition serves as a major control mechanism in biological systems, typified by the regulation of allosteric enzymes. In addition, many drugs and toxic agents act by inhibiting enzymes. This type of enzyme inhibition is not usually the result of evolutionary forces, as it is for allosteric enzymes, but rather due to design of inhibitors by scientists or simple chance discovery of inhibitory molecules. Examining inhibition can be a source of insight into the mechanism of enzyme action: specific inhibitors can often be used to identify residues critical for catalysis. Transition-state analogs are especially potent inhibitors.
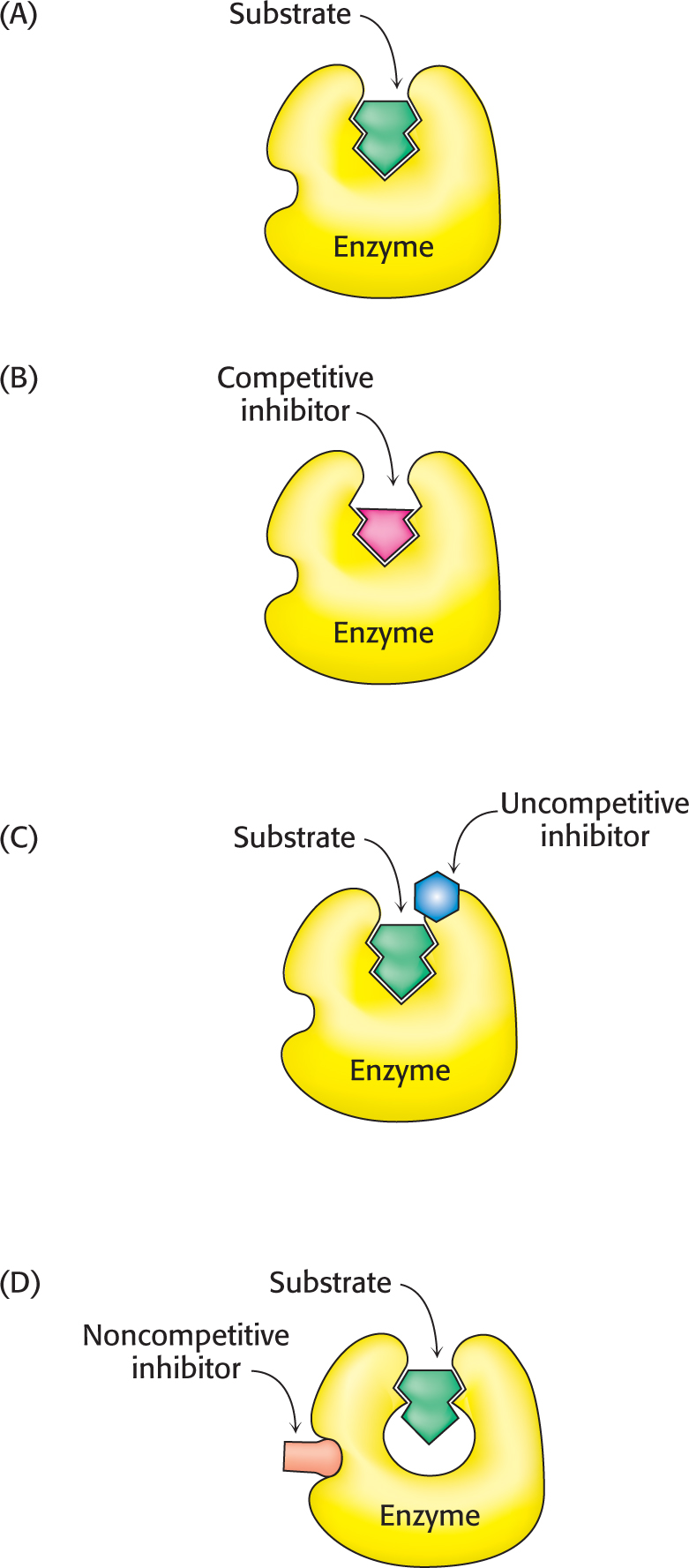
Figure 8.6: Reversible inhibitors. (A) Substrate binds to an enzyme’s active site to form an enzyme–substrate complex. (B) A competitive inhibitor binds at the active site and thus prevents the substrate from binding. (C) An uncompetitive inhibitor binds only to the enzyme–substrate complex. (D) A noncompetitive inhibitor does not prevent the substrate from binding.
Enzyme inhibition can be either reversible or irreversible. We begin the investigation of enzyme inhibition by first examining reversible inhibition. In contrast with irreversible inhibition, reversible inhibition is characterized by rapid dissociation of the enzyme–inhibitor complex. There are three common types of reversible inhibition: competitive inhibition, uncompetitive inhibition, and noncompetitive inhibition. These three types of inhibition differ in the nature of the interaction between the enzyme and the inhibitor and in the inhibitor’s effect on enzyme kinetics (Figure 8.6). We will consider each of these types in turn.
In competitive inhibition, the inhibitor resembles the substrate and binds to the active site of the enzyme (Figure 8.6B). The substrate is thereby prevented from binding to the same active site. An enzyme can bind substrate (forming an ES complex) or inhibitor (EI), but not both (ESI). A competitive inhibitor diminishes the rate of catalysis by reducing the proportion of enzyme molecules bound to a substrate. At any given inhibitor concentration, competitive inhibition can be relieved by increasing the substrate concentration. Under these conditions, the substrate “outcompetes” the inhibitor for the active site.
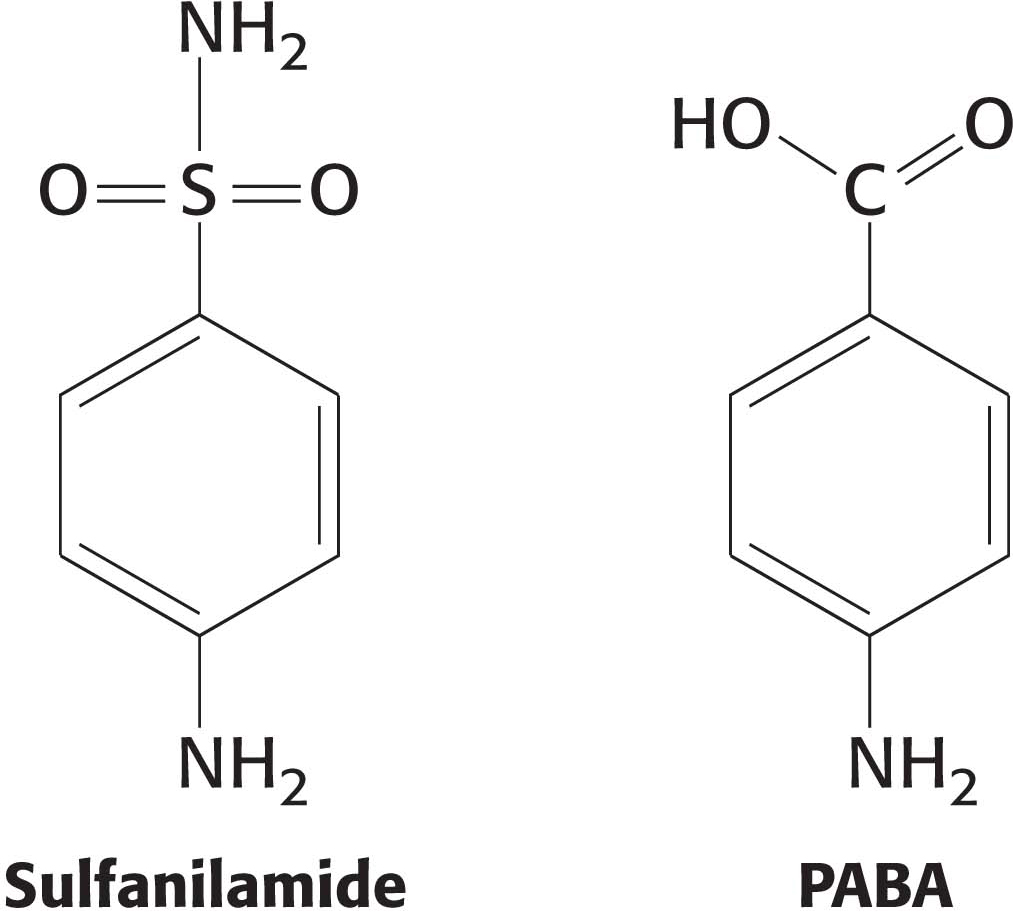
Some competitive inhibitors are useful drugs. One of the earliest examples was the use of sulfanilamide as an antibiotic. Sulfanilamide is an example of a sulfa drug, a sulfur-containing antibiotic. Structurally, sulfanilamide mimics p-aminobenzoic acid (PABA), a metabolite required by bacteria for the synthesis of the coenzyme folic acid. Sulfanilamide binds to the enzyme that normally metabolizes PABA and competitively inhibits it, preventing folic acid synthesis. Human beings, unlike bacteria, absorb folic acid from the diet and are thus unaffected by the sulfa drug. Other competitive inhibitors commonly used as drugs include ibuprofen, which inhibits a cyclooxygenase that helps to generate the inflammatory response, and statins, which inhibit the key enzyme in cholesterol synthesis.
Uncompetitive inhibition is substrate-dependent inhibition in that the inhibitor binds only to the enzyme–substrate complex. The uncompetitive inhibitor’s binding site is created only when the enzyme binds the substrate (Figure 8.6C). Uncompetitive inhibition cannot be overcome by the addition of more substrate. The herbicide glyphosate, also known as Roundup, is an uncompetitive inhibitor of an enzyme in the biosynthetic pathway for aromatic amino acids in plants. The plant dies because it lacks these amino acids.
In noncompetitive inhibition, the inhibitor and substrate can bind simultaneously to an enzyme molecule at different binding sites (Figure 8.6D). A noncompetitive inhibitor acts by decreasing the overall number of active enzyme molecules rather than by diminishing the proportion of enzyme molecules that are bound to substrate. Noncompetitive inhibition, in contrast with competitive inhibition, cannot be overcome by increasing the substrate concentration. Doxycycline, an antibiotic, functions at low concentrations as a noncompetitive inhibitor of a bacterial proteolytic enzyme (collagenase). Inhibition of this enzyme prevents the growth and reproduction of bacteria that cause gum (periodontal) disease.
Reversible Inhibitors Are Kinetically Distinguishable
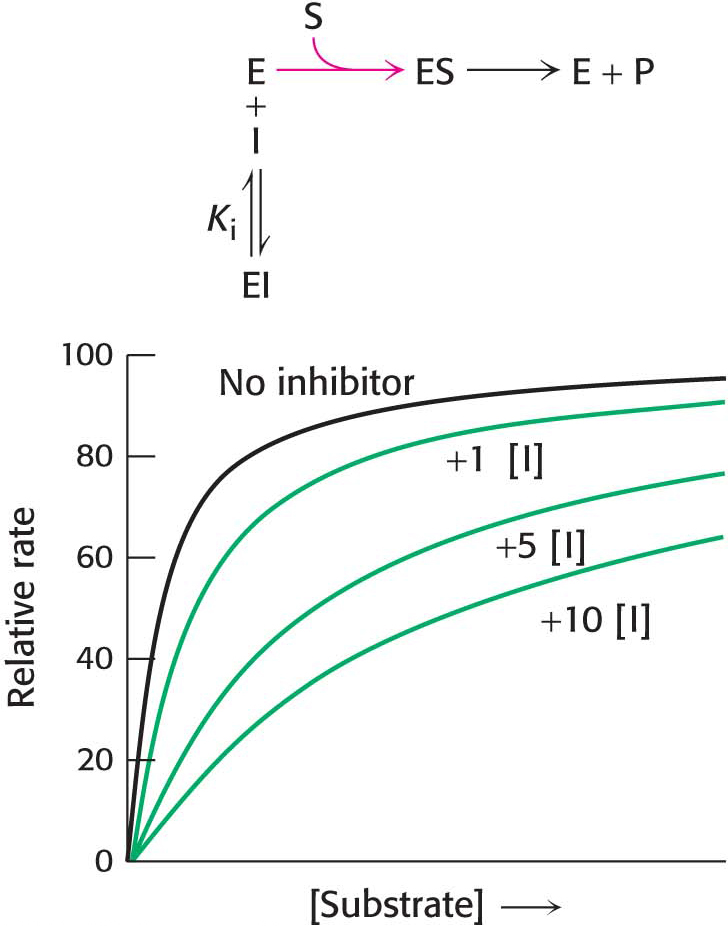
Figure 8.7: Kinetics of a competitive inhibitor. As the concentration of a competitive inhibitor increases, higher concentrations of substrate are required to attain a particular reaction velocity. The reaction pathway suggests how sufficiently high concentrations of substrate can completely relieve competitive inhibition.
How can we determine whether a reversible inhibitor acts by competitive, uncompetitive, or noncompetitive inhibition? Let us consider only enzymes that exhibit Michaelis–Menten kinetics—that is, enzymes that are not allosterically inhibited. Measurements of the rates of catalysis at different concentrations of substrate and inhibitor serve to distinguish the three types of reversible inhibition. In competitive inhibition, the inhibitor competes with the substrate for the active site. The hallmark of competitive inhibition is that it can be overcome by a sufficiently high concentration of substrate (Figure 8.7). The effect of a competitive inhibitor is to increase the apparent value of KM, meaning that more substrate is needed to obtain the same reaction rate. This new apparent value of KM is called 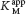 . In the presence of a competitive inhibitor, an enzyme will have the same Vmax as in the absence of an inhibitor. At a sufficiently high concentration, virtually all the active sites are filled by substrate, and the enzyme is fully operative. The more inhibitor present, the more substrate is required to displace it and reach Vmax.
. In the presence of a competitive inhibitor, an enzyme will have the same Vmax as in the absence of an inhibitor. At a sufficiently high concentration, virtually all the active sites are filled by substrate, and the enzyme is fully operative. The more inhibitor present, the more substrate is required to displace it and reach Vmax.
In uncompetitive inhibition, the inhibitor binds only to the ES complex. This enzyme–substrate–inhibitor complex, ESI, does not proceed to form any product. Because some unproductive ESI complex will always be present, Vmax will be lower in the presence of an inhibitor than in its absence (Figure 8.8). The uncompetitive inhibitor also lowers the apparent value of KM, because the inhibitor binds to ES to form ESI, depleting ES. To maintain the equilibrium between E and ES, more S binds to E, increasing the apparent value of k1 and thereby reducing the apparent value of KM (see equation 8 in Section 7.2). Thus, a lower concentration of S is required to form half of the maximal concentration of ES, resulting in a reduction of the apparent value of KM, now called 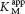 . Likewise, the value of Vmax is decreased to a new value called
. Likewise, the value of Vmax is decreased to a new value called 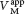 .
.
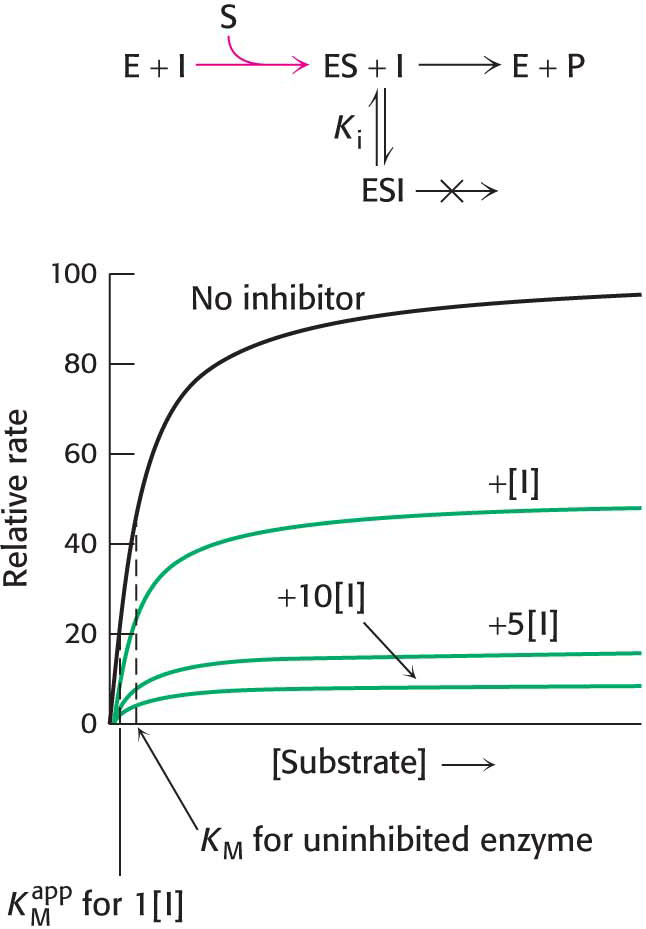
Figure 8.8: Kinetics of an uncompetitive inhibitor. The reaction pathway shows that the inhibitor binds only to the enzyme–substrate complex. Consequently, Vmax cannot be attained, even at high substrate concentrations. Notice that the apparent value for 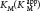 is lowered, becoming smaller as more inhibitor is added.
is lowered, becoming smaller as more inhibitor is added.
In noncompetitive inhibition (Figure 8.9), a substrate can bind to the enzyme–inhibitor complex as well as to the enzyme alone. In either case, the enzyme–inhibitor–substrate complex does not proceed to form product. The value of Vmax is decreased to the new value 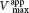 , whereas the value of KM is unchanged. Why is Vmax lowered although KM remains unchanged? In essence, the inhibitor simply lowers the concentration of functional enzyme. The resulting solution behaves as a more dilute solution of enzyme. Noncompetitive inhibition cannot be overcome by increasing the substrate concentration.
, whereas the value of KM is unchanged. Why is Vmax lowered although KM remains unchanged? In essence, the inhibitor simply lowers the concentration of functional enzyme. The resulting solution behaves as a more dilute solution of enzyme. Noncompetitive inhibition cannot be overcome by increasing the substrate concentration.
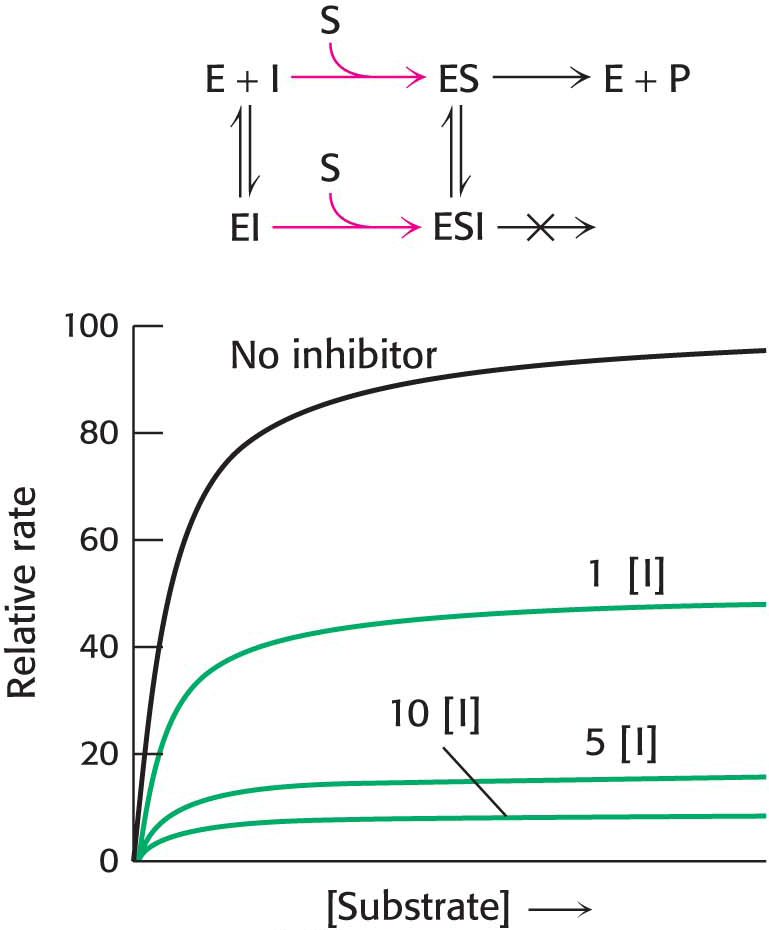
Figure 8.9: Kinetics of a noncompetitive inhibitor. The reaction pathway shows that the inhibitor binds both to free enzyme and to the enzyme–substrate complex. Consequently, as with uncompetitive competition, Vmax cannot be attained. KM remains unchanged, and so the reaction rate increases more slowly at low substrate concentrations than is the case for uncompetitive competition.
Double-reciprocal plots are especially useful for distinguishing competitive, uncompetitive, and noncompetitive inhibitors. In competitive inhibition, the intercept on the y axis, 1/Vmax, is the same in the presence and in the absence of inhibitor, although the slope (KM/Vmax) is increased (Figure 8.10). The intercept is unchanged because a competitive inhibitor does not alter Vmax. The increase in the slope of the 1/V0 versus 1/[S] plot indicates the strength of binding of competitive inhibitor.
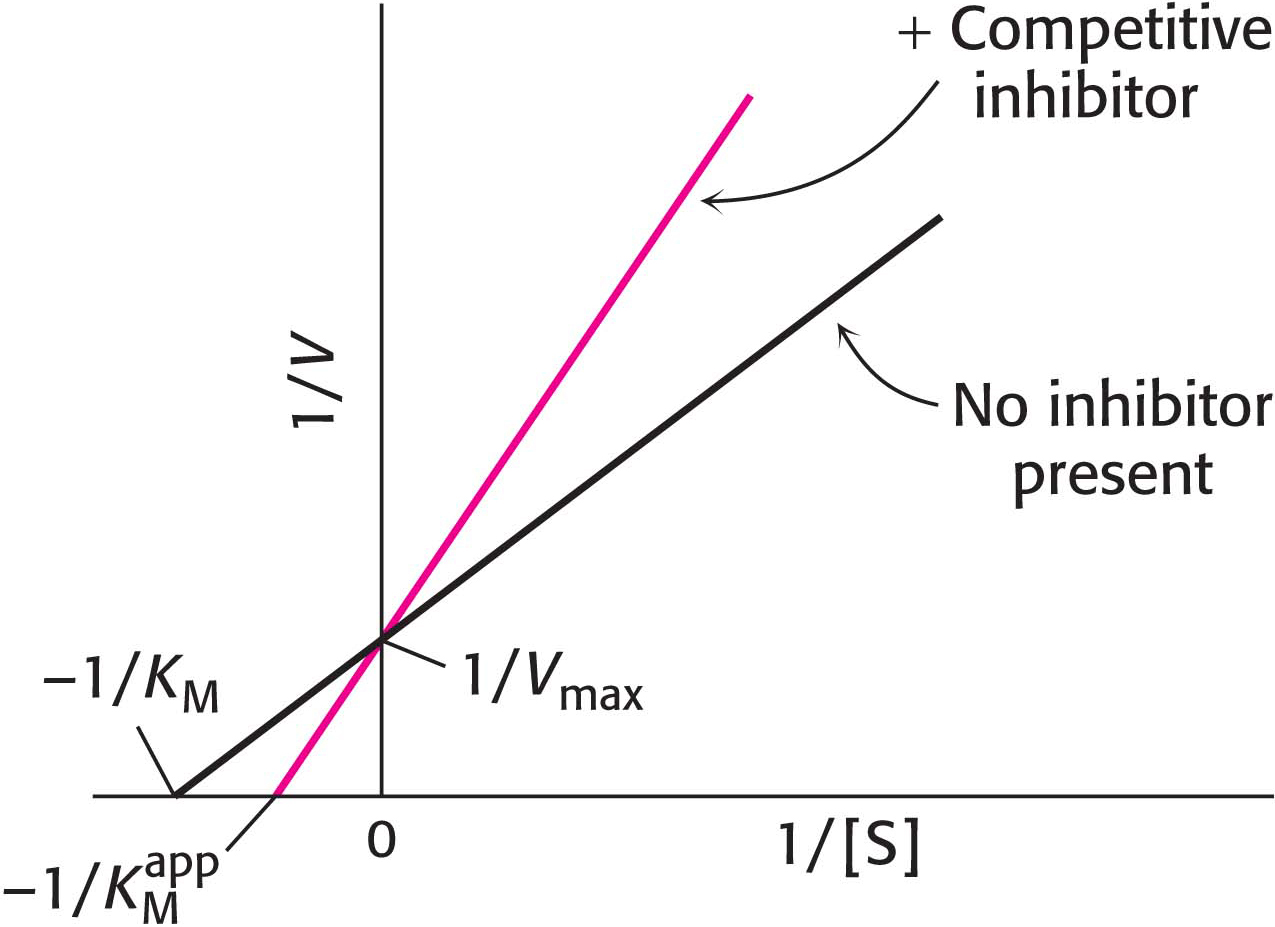
Figure 8.10: Competitive inhibition illustrated on a double-reciprocal plot. A double-reciprocal plot of enzyme kinetics in the presence and absence of a competitive inhibitor illustrates that the inhibitor has no effect on Vmax but increases KM.
In uncompetitive inhibition (Figure 8.11), the inhibitor combines only with the enzyme–substrate complex. The slope of the line is the same as that for the uninhibited enzyme, but the intercept on the y axis, 1/Vmax, is increased. Consequently, for uncompetitive inhibition, the lines in double-reciprocal plots are parallel.
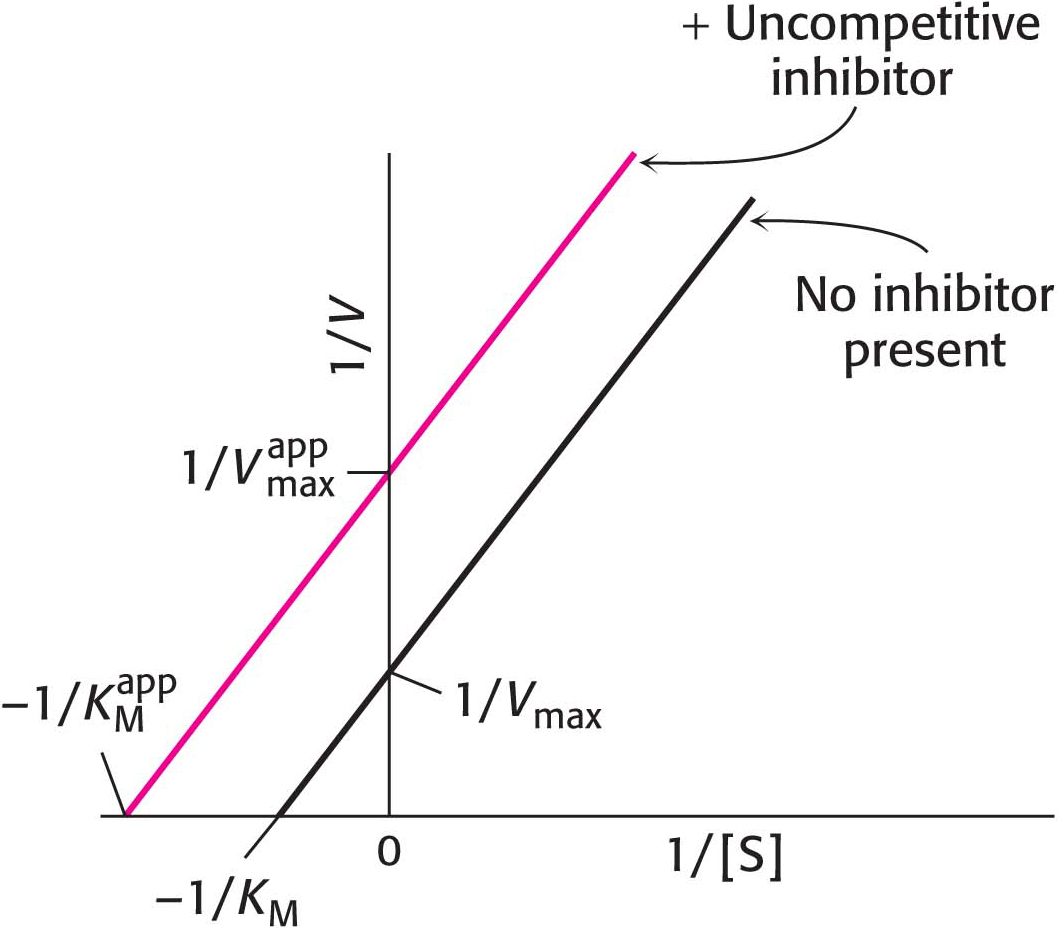
Figure 8.11: Uncompetitive inhibition illustrated on a double-reciprocal plot. An uncompetitive inhibitor does not affect the slope of the double-reciprocal plot. Vmax and KM are reduced by equivalent amounts.
In noncompetitive inhibition (Figure 8.12), the inhibitor can combine with either the enzyme or the enzyme–substrate complex. In pure noncompetitive inhibition, the values of the dissociation constants of the inhibitor and enzyme and of the inhibitor and enzyme–substrate complex are equal. The value of Vmax is decreased to the new value 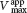 , and so the intercept on the vertical axis is increased. The slope when the inhibitor is present, which is equal to
, and so the intercept on the vertical axis is increased. The slope when the inhibitor is present, which is equal to 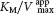 , is larger by the same factor. In contrast with Vmax, KM is not affected by pure noncompetitive inhibition.
, is larger by the same factor. In contrast with Vmax, KM is not affected by pure noncompetitive inhibition.
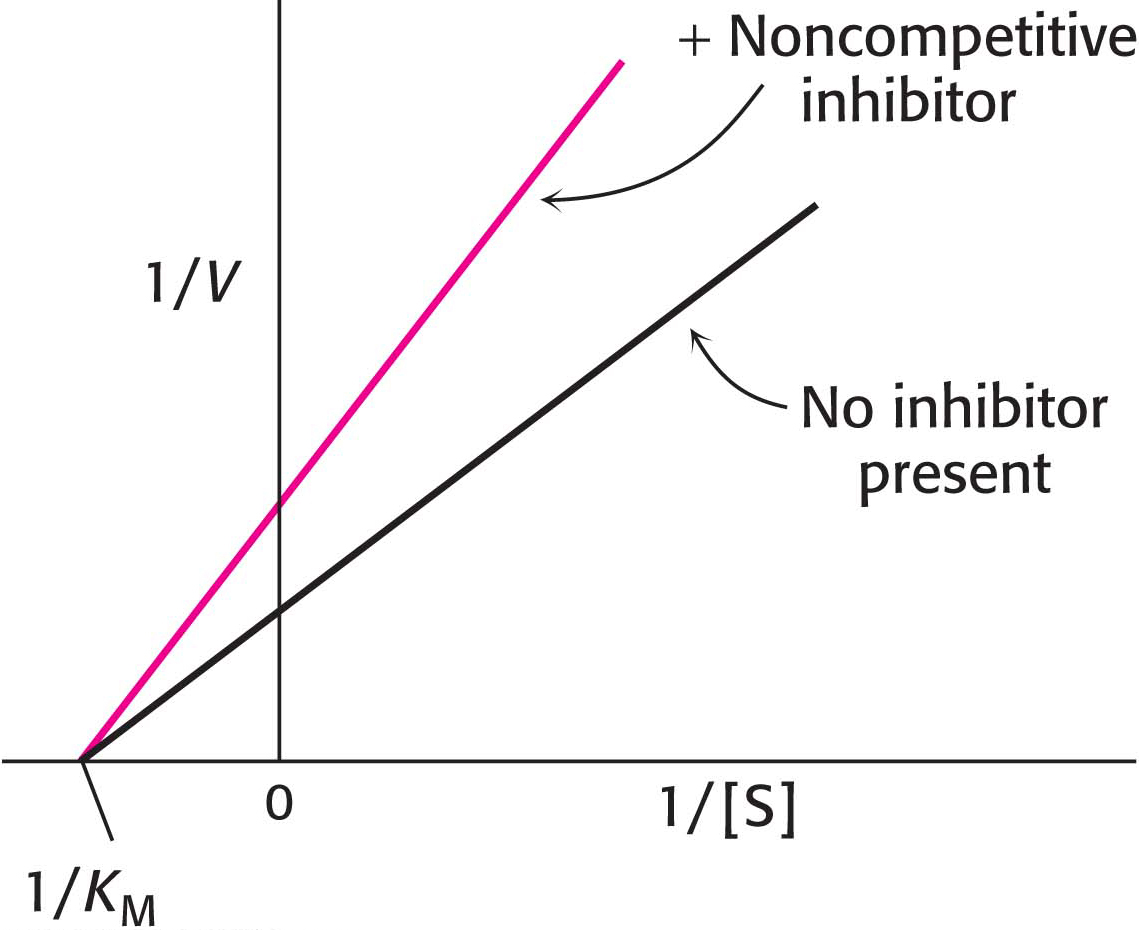
Figure 8.12: Noncompetitive inhibition illustrated on a double-reciprocal plot. A double-reciprocal plot of enzyme kinetics in the presence and absence of a noncompetitive inhibitor shows that KM is unaltered and Vmax is decreased.
Irreversible Inhibitors Can Be Used to Map the Active Site
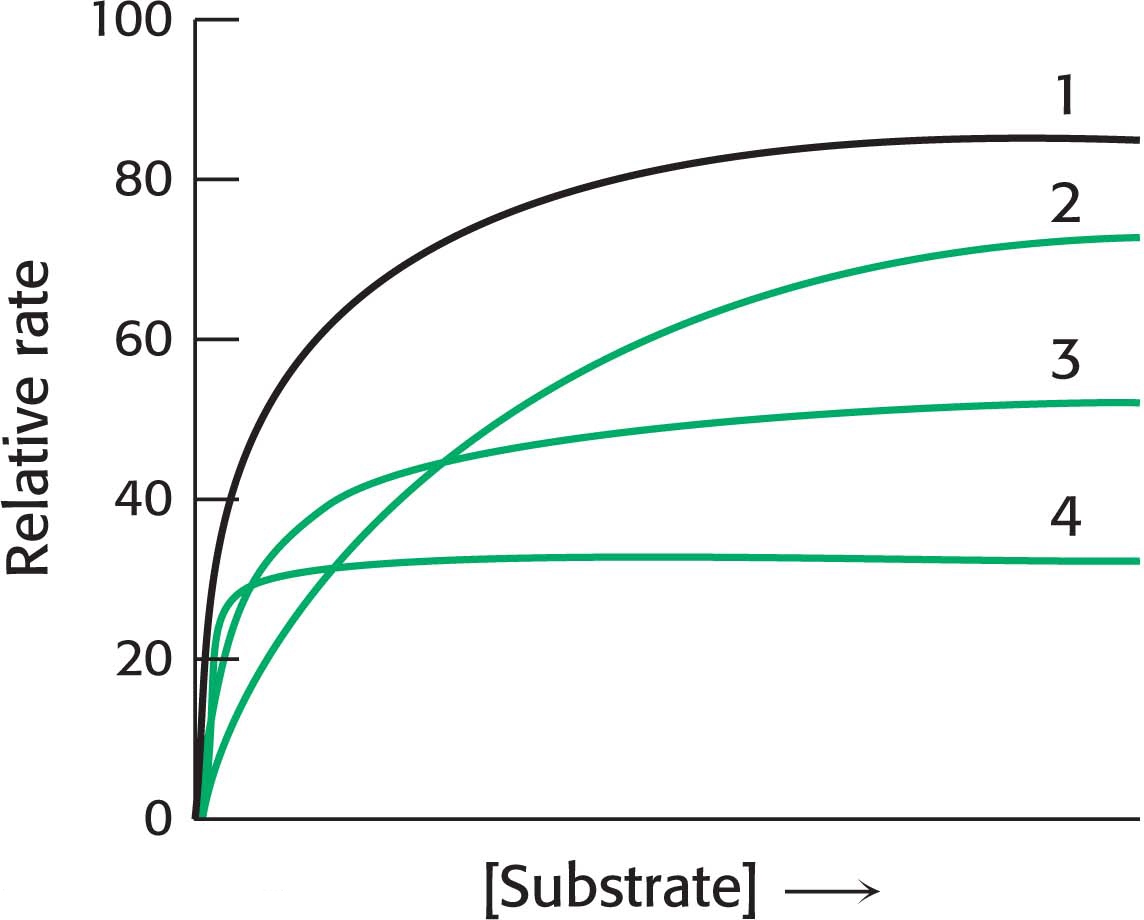
!quickquiz! QUICK QUIZ 1
In the following graph, identify the curve that corresponds to each of the following conditions: no inhibition, competitive inhibition, noncompetitive inhibition, uncompetitive inhibition.
Whereas a reversible inhibitor will both bind to an enzyme and dissociate from it rapidly, an irreversible inhibitor dissociates very slowly from its target enzyme because it has become tightly bound to the enzyme, either covalently or noncovalently. Some irreversible inhibitors are important drugs. Penicillin acts by covalently modifying the enzyme transpeptidase, thereby preventing the synthesis of bacterial cell walls and thus killing the bacteria. Aspirin acts by covalently modifying the enzyme cyclooxygenase (the same enzyme inhibited by ibuprofen), reducing the synthesis of inflammatory signals.
Irreversible inhibitors that covalently bind to an enzyme are tools for elucidating the mechanism of enzymes. The first step in determining the chemical mechanism of an enzyme is to determine which functional groups are required for enzyme activity. Irreversible inhibitors modify functional groups, which can then be identified. If treatment with an irreversible inhibitor results in a loss of enzyme activity, then this loss suggests that the modified group is required for enzyme activity. Irreversible inhibitors can be assorted into four categories: group-specific reagents, affinity labels (substrate analogs), suicide inhibitors, and transition-state analogs.
Group-specific reagents modify specific R groups of amino acids. An example of a group-specific reagent is diisopropylphosphofluoridate (DIPF). DIPF inhibits the proteolytic enzyme chymotrypsin by modifying only 1 of the 28 serine residues in the protein, implying that this serine residue is especially reactive. As we will see on page 141, this serine residue is indeed at the active site. DIPF also revealed a reactive serine residue in acetylcholinesterase, an enzyme important in the transmission of nerve impulses (Figure 8.13). Thus, DIPF and similar compounds that bind and inactivate acetylcholinesterase are potent nerve gases.
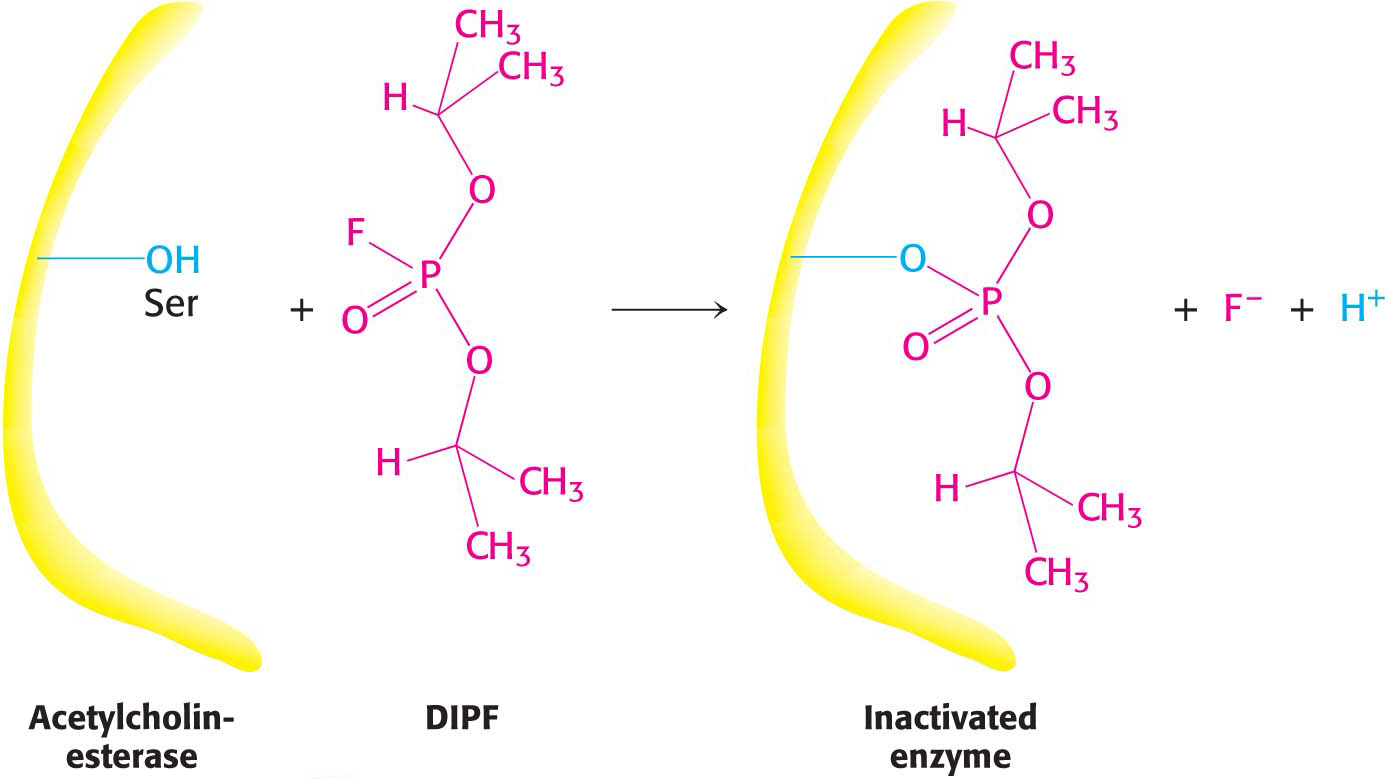
Figure 8.13: Enzyme inhibition by diisopropylphosphofluoridate (DIPF), a group-specific reagent. DIPF can inhibit an enzyme by covalently modifying a crucial serine residue.
Affinity labels, also called substrate analogs, are molecules that covalently modify active-site residues and are structurally similar to an enzyme’s substrate. They are thus more specific for an enzyme’s active site than are group-specific reagents. Tosyl-l-phenylalanine chloromethyl ketone (TPCK) is an affinity label for chymotrypsin (Figure 8.14). TPCK binds at the active site and then reacts irreversibly with a histidine residue at that site, inhibiting the enzyme.
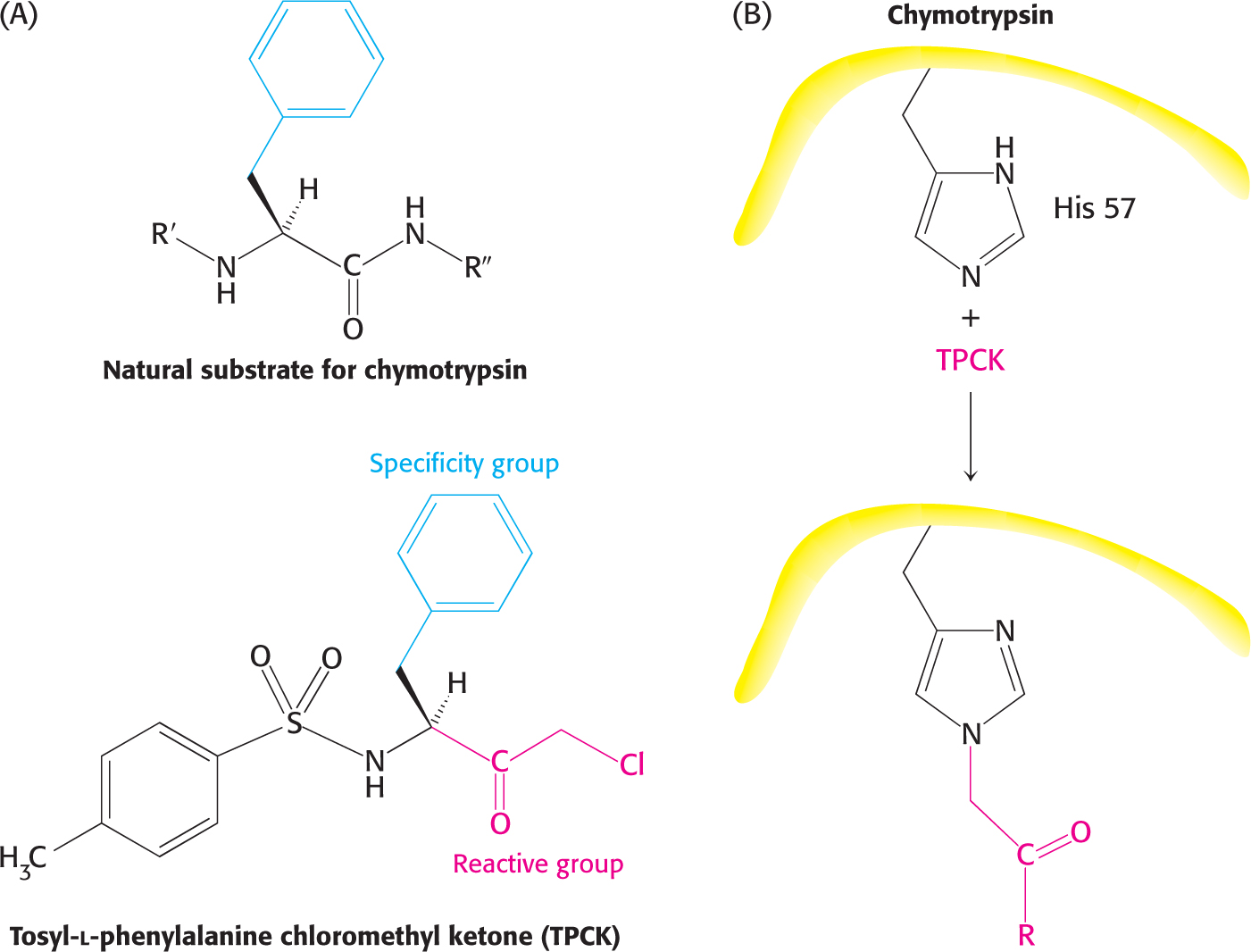
Figure 8.14: Affinity labeling. (A) Tosyl-l-phenylalanine chloromethyl ketone (TPCK) is a reactive analog of the normal substrate for the enzyme chymotrypsin. (B) TPCK binds at the chymotrypsin active site and modifies an essential histidine residue.
Suicide inhibitors, or mechanism-based inhibitors, are chemically modified substrates. These molecules provide researchers with the most specific means of modifying an enzyme’s active site. The inhibitor binds to the enzyme as a substrate and is initially processed by the normal catalytic mechanism. The mechanism of catalysis then generates a chemically reactive intermediate that inactivates the enzyme through covalent modification. The fact that the enzyme participates in its own irreversible inhibition strongly suggests that the covalently modified group on the enzyme is catalytically vital. The antibiotic penicillin is a suicide inhibitor of the enzyme that synthesizes bacterial cell walls.
Transition-state analogs are potent inhibitors of enzymes. As discussed earlier, the formation of the transition state is crucial to enzyme catalysis. An important piece of evidence supporting the role of transition-state formation in catalysis is the inhibitory power of transition-state analogs.
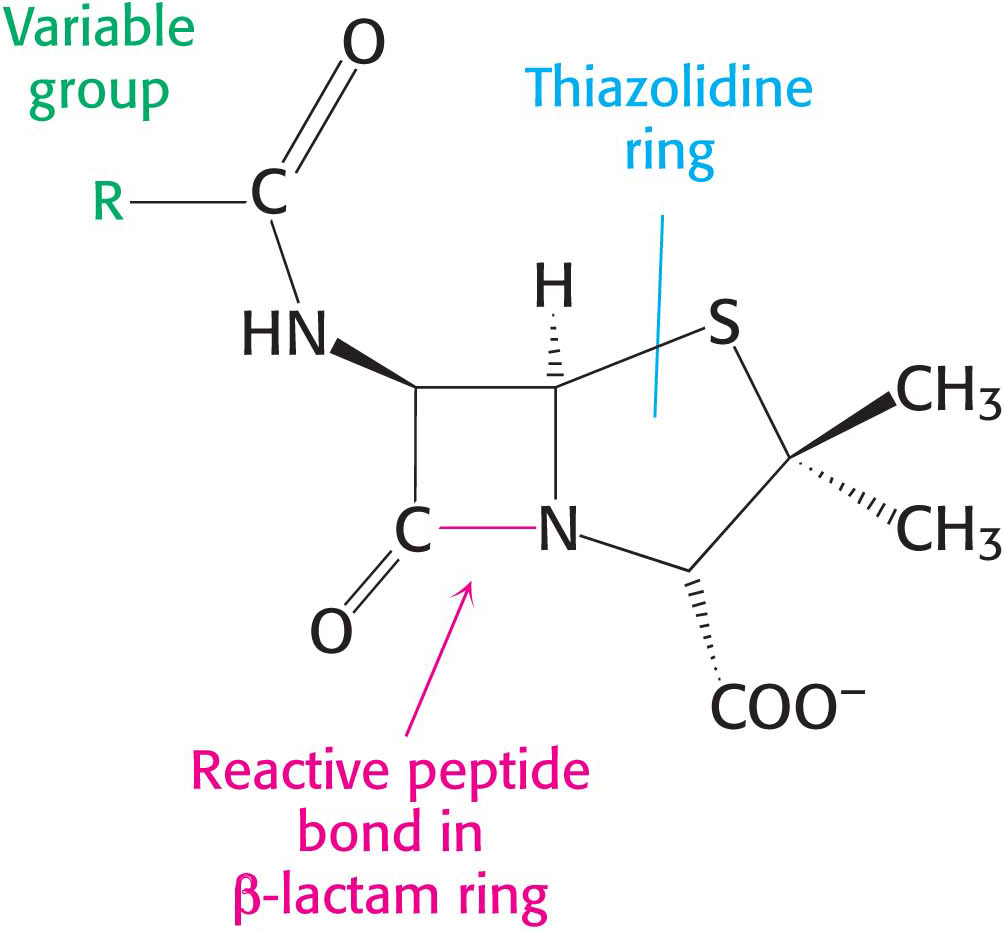
Figure 8.15: The structure of penicillin. The reactive site of penicillin is the peptide bond of its β-lactam ring.
!clinic! CLINICAL INSIGHT: Penicillin Irreversibly Inactivates a Key Enzyme in Bacterial Cell-Wall Synthesis
Penicillin, the first antibiotic discovered, consists of a thiazolidine ring fused to a β-lactam ring to which a variable R group is attached by a peptide bond (Figure 8.15). This structure can undergo a variety of rearrangements, and, in particular, the β-lactam ring is very unstable. Indeed, this instability is closely tied to the antibiotic action of penicillin, as will be evident shortly.
How does penicillin inhibit bacterial growth? Let us consider the bacterium Staphylococcus aureus, the most common cause of staph infections. Penicillin works by interfering with the synthesis of the S. aureus cell walls. The S. aureus cell wall is made up of a macromolecule, called a peptidoglycan (Figure 8.16), which consists of linear polysaccharide chains that are cross-linked by short peptides (pentaglycines and tetrapeptides). The enormous bag-shaped peptidoglycan confers mechanical support and prevents bacteria from bursting in response to their high internal osmotic pressure. Glycopeptide transpeptidase catalyzes the formation of the cross-links that make the peptidoglycan so stable (Figure 8.17). Bacterial cell walls are distinctive in containing D amino acids, which form cross-links by a mechanism different from that used to synthesize proteins.
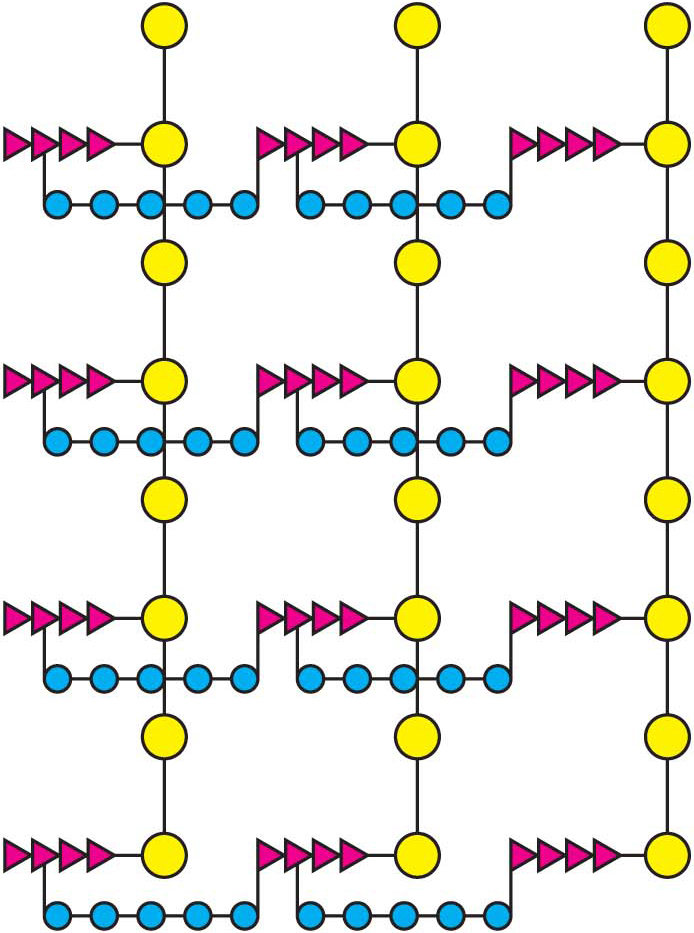
Figure 8.16: A schematic representation of the peptidoglycan in Staphylococcus aureus. The sugars are shown in yellow, the tetrapeptides in red, and the pentaglycine bridges in blue. The cell wall is a single, enormous, bag-shaped macromolecule because of extensive cross-linking.
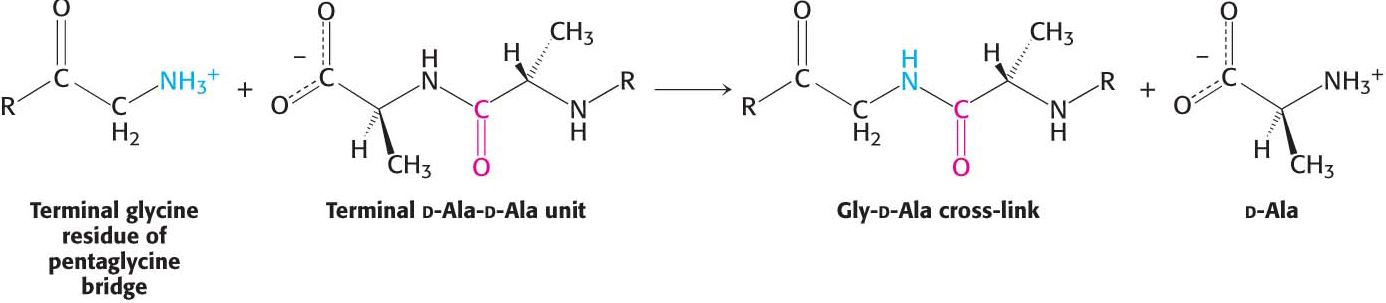
Figure 8.17: The formation of cross-links in S. aureus peptidoglycan. The terminal amino group of the pentaglycine bridge in the cell wall attacks the peptide bond between two d-alanine residues to form a cross-link.
Penicillin inhibits the cross-linking transpeptidase by the Trojan horse stratagem: it mimics a normal substrate to enter the active site. To create cross-links between the tetrapeptides and pentaglycines, the transpeptidase normally forms an acyl intermediate with the penultimate d-alanine residue of the tetrapeptide. This covalent acyl-enzyme intermediate then reacts with the amino group of the terminal glycine in another peptide to form the cross-link (Figure 8.18). Penicillin is welcomed into the active site of the transpeptidase because it mimics the d-Ala-d-Ala moiety of the normal substrate. On binding to the transpeptidase, the serine residue at the active site attacks the carbonyl carbon atom of the lactam ring to form the penicilloyl-serine derivative (Figure 8.19). This penicilloyl-enzyme does not react further. Hence, the transpeptidase is irreversibly inhibited and cell-wall synthesis cannot take place. Because the peptidase participates in its own inactivation, penicillin acts as a suicide inhibitor.
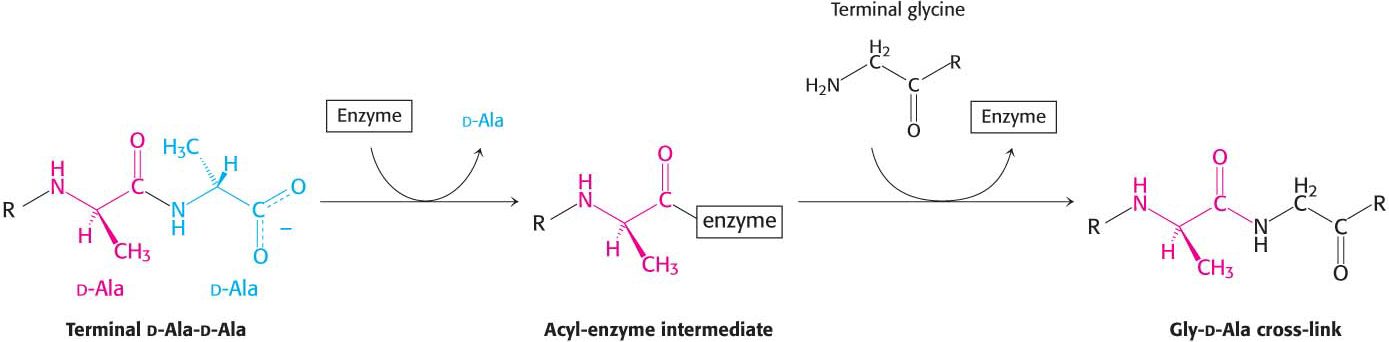
Figure 8.18: A transpeptidation reaction. An acyl-enzyme intermediate is formed in the transpeptidation reaction leading to cross-link formation.
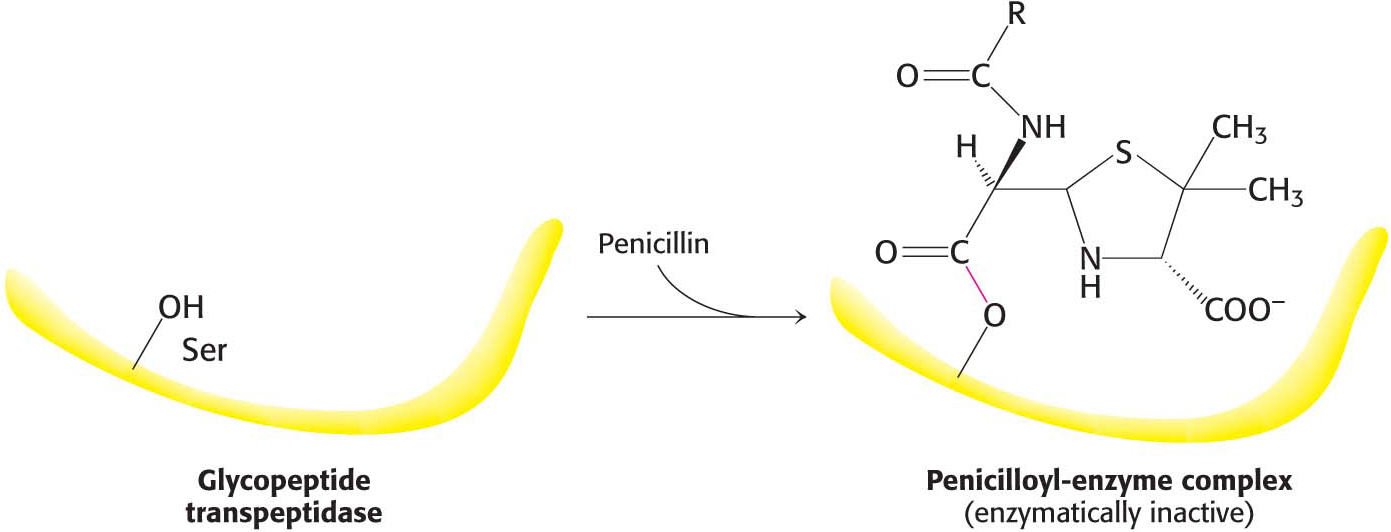
Figure 8.19: The formation of a penicilloyl-enzyme complex. Penicillin reacts with the transpeptidase to form an inactive complex, which is indefinitely stable.