APPENDIX: Visualizing Molecular Structures II: Proteins
APPENDIX: Visualizing Molecular Structures II: Proteins
Scientists have developed powerful techniques for the determination of protein structures, as will be considered in Chapter 3. In most cases, these techniques allow the positions of the thousands of atoms within a protein structure to be determined. The final results from such an experiment include the x, y, and z coordinates for each atom in the structure. These coordinate files are compiled in the Protein Data Bank (http:/
Space-Filling Models
Space-
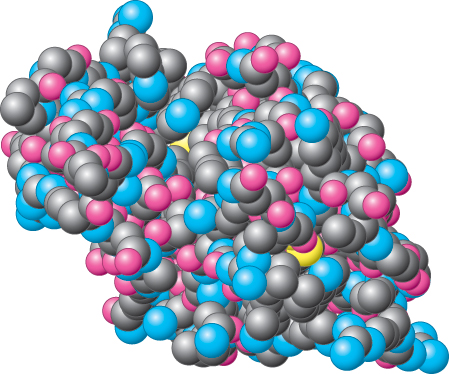
Space-
Ball-and-Stick Models
Ball-
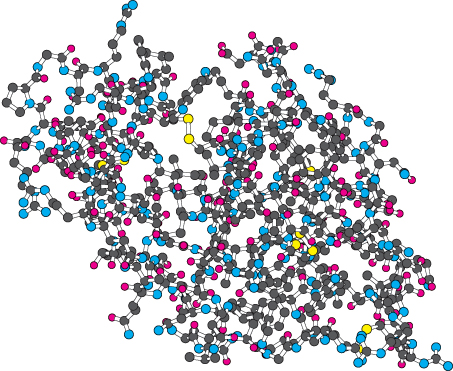
Because space-
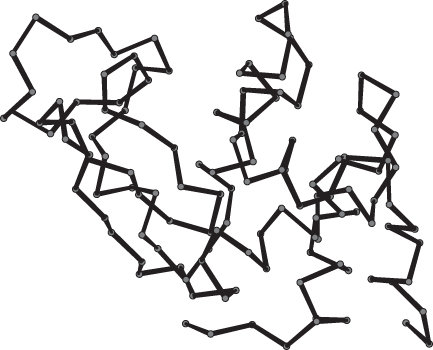
Backbone Models
Backbone models show only the backbone atoms of a polypeptide chain or even only the α-carbon atom of each amino acid. Atoms are linked by lines representing bonds; if only α-carbon atoms are depicted, lines connect α-carbon atoms of amino acids that are adjacent in the amino acid sequence (Figure 2.68). In this book, backbone models show only the lines connecting the α-carbon atoms; other carbon atoms are not depicted.
62
A backbone model shows the overall course of the polypeptide chain much better than a space-
Ribbon Diagrams
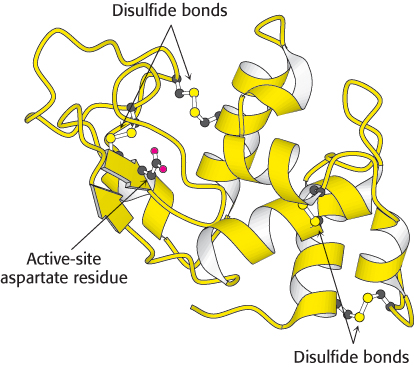
Ribbon diagrams are highly schematic and most commonly used to accent a few dramatic aspects of protein structure, such as the α helix (depicted as a coiled ribbon or a cylinder), the β strand (a broad arrow), and loops (thin tubes), to provide clear views of the folding patterns of proteins (Figure 2.69). The ribbon diagram allows the course of a polypeptide chain to be traced and readily shows the secondary structural elements. Thus, ribbon diagrams of proteins that are related to one another by evolutionary divergence appear similar (Figure 6.15), whereas unrelated proteins are clearly distinct.
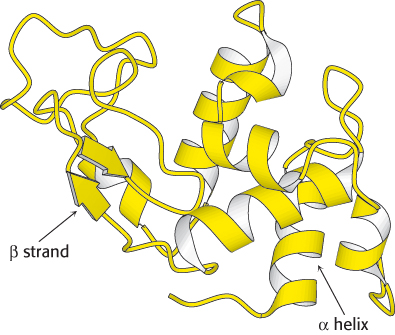
In this book, coiled ribbons will be generally used to depict α helices. However, for membrane proteins, which are often quite complex, cylinders will be used rather than coiled ribbons. This convention will also make membrane proteins with their membrane-
Bear in mind that the open appearance of ribbon diagrams is deceptive. As noted earlier, protein structures are tightly packed and have little open space. The openness of ribbon diagrams makes them particularly useful as frameworks in which to highlight additional aspects of protein structure. Active sites, substrates, bonds, and other structural fragments can be included in ball-