Ethanol metabolism leads to an excess of NADH
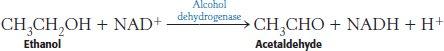
Ethanol cannot be excreted and must be metabolized, primarily by the liver. This metabolism is accomplished by two pathways. The first pathway comprises two steps. The first step, catalyzed by the enzyme alcohol dehydrogenase, takes place in the cytoplasm:
The second step, catalyzed by aldehyde dehydrogenase, takes place in mitochondria:
Note that ethanol consumption leads to an accumulation of NADH. This high concentration of NADH inhibits gluconeogenesis by preventing the oxidation of lactate to pyruvate. In fact, the high concentration of NADH will cause the reverse reaction to predominate, and lactate will accumulate. The consequences may be hypoglycemia and lactic acidosis.
The overabundance of NADH also inhibits fatty acid oxidation. An important metabolic purpose of fatty acid oxidation is to generate NADH for ATP generation by oxidative phosphorylation, but an alcohol consumer’s NADH needs are met by ethanol metabolism. In fact, the excess NADH signals that conditions are right for fatty acid synthesis. Hence, triacylglycerols accumulate in the liver, leading to a condition known as “fatty liver” that is exacerbated in obese persons. The biochemical effects of ethanol consumption can be quite rapid. For instance, fat accumulates in the liver within a few days of moderate alcohol consumption. This accumulation is reversible with a decrease in alcohol intake.
The second pathway for ethanol metabolism uses cytochrome P450 enzymes. This means of ethanol metabolism is also called the microsomal ethanol-oxidizing system (MEOS). This cytochrome P450-dependent pathway (Section 26.4) generates acetaldehyde and subsequently acetate while oxidizing biosynthetic reducing power, NADPH, to NADP+. Because it uses oxygen, this pathway generates free radicals that damage tissues. Moreover, because the system consumes NADPH, the antioxidant glutathione cannot be regenerated (Section 20.5), exacerbating the oxidative stress.
What are the effects of the other metabolites of ethanol? Liver mitochondria can convert acetate into acetyl CoA in a reaction requiring ATP. The enzyme is the thiokinase that normally activates short-chain fatty acids.
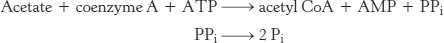
However, further processing of the acetyl CoA by the citric acid cycle is blocked, because NADH inhibits two important citric acid cycle regulatory enzymes—isocitrate dehydrogenase and α-ketoglutarate dehydrogenase. The accumulation of acetyl CoA has several consequences. First, ketone bodies will form and be released into the blood, aggravating the acidic condition already resulting from the high lactate concentration. The processing of the acetate in the liver becomes inefficient, leading to a buildup of acetaldehyde. This very reactive compound forms covalent bonds with many important functional groups in proteins, impairing protein function. If ethanol is consistently consumed at high levels, the acetaldehyde can significantly damage the liver, eventually leading to cell death.
Liver damage from excessive ethanol consumption occurs in three stages. The first stage is the aforementioned development of fatty liver. In the second stage—alcoholic hepatitis—groups of cells die and inflammation results. This stage can itself be fatal. In stage three—cirrhosis—fibrous structure and scar tissue are produced around the dead cells. Cirrhosis impairs many of the liver’s biochemical functions. The cirrhotic liver is unable to convert ammonia into urea, and blood levels of ammonia rise. Ammonia is toxic to the nervous system and can cause coma and death. Cirrhosis of the liver arises in about 25% of alcoholics, and about 75% of all cases of liver cirrhosis are the result of alcoholism. Viral hepatitis is a non-alcoholic cause of liver cirrhosis.
Excess ethanol consumption disrupts vitamin metabolism
The adverse effects of ethanol are not limited to the metabolism of ethanol itself. Vitamin A (retinol) is converted into retinoic acid, an important signal molecule for growth and development in vertebrates, by the same dehydrogenases that metabolize ethanol. Consequently, this activation does not take place in the presence of ethanol, which acts as a competitive inhibitor. Moreover, the P450 enzymes induced by ethanol inactivate retinoic acid. These disruptions in the retinoic acid signaling pathway are believed to be responsible, at least in part, for fetal alcohol syndrome as well as the development of a variety of cancers.
The disruption of vitamin A metabolism is a direct result of the biochemical changes induced by excess ethanol consumption. Other disruptions in metabolism result from another common characteristic of alcoholics—malnutrition. Alcoholics will frequently drink instead of eating. A dramatic neurological disorder, referred to as Wernicke–Korsakoff syndrome, results from insufficient intake of the vitamin thiamine. Symptoms include mental confusion, unsteady gait, and lack of fine motor skills. The symptoms of Wernicke–Korsakoff syndrome are similar to those of beriberi (Section 17.4) because both conditions result from a lack of thiamine. Thiamine is converted into the coenzyme thiamine pyrophosphate, a key constituent of the pyruvate dehydrogenase complex. Recall that this complex links glycolysis with the citric acid cycle. Disruptions in the pyruvate dehydrogenase complex are most evident as neuromuscular disorders because the nervous system is normally dependent on glucose for energy generation.
Alcoholic scurvy is occasionally observed because of an insufficient ingestion of vitamin C. Vitamin C is required for the formation of stable collagen fibers. The symptoms of scurvy include skin lesions and blood-vessel fragility. Most notable are bleeding gums, the loss of teeth, and periodontal infections. Gums are especially sensitive to a lack of vitamin C because the collagen in gums turns over rapidly. What is the biochemical basis for scurvy? Vitamin C is required for the synthesis of 4-hydroxyproline, an amino acid necessary for collagen stability. To form this unusual amino acid, proline residues on the amino side of glycine residues in nascent collagen chains become hydroxylated. One oxygen atom from O2 becomes attached to C-4 of proline while the other oxygen atom is taken up by α-ketoglutarate, which is converted into succinate (Figure 27.16). This reaction is catalyzed by prolyl hydroxylase, a dioxygenase, which requires an Fe2+ ion to activate O2. The enzyme also converts α-ketoglutarate into succinate without hydroxylating proline. In this partial reaction, an oxidized iron complex is formed, which inactivates the enzyme. How is the active enzyme regenerated? Ascorbate (vitamin C) comes to the rescue by reducing the ferric ion of the inactivated enzyme. In the recovery process, ascorbate is oxidized to dehydroascorbic acid (Figure 27.17). Thus, ascorbate serves here as a specific antioxidant. Why does impaired hydroxylation have such devastating consequences? Collagen synthesized in the absence of ascorbate is less stable than the normal protein. Hydroxyproline stabilizes the collagen triple helix by forming interstrand hydrogen bonds. The abnormal fibers formed by insufficiently hydroxylated collagen account for the symptoms of scurvy.
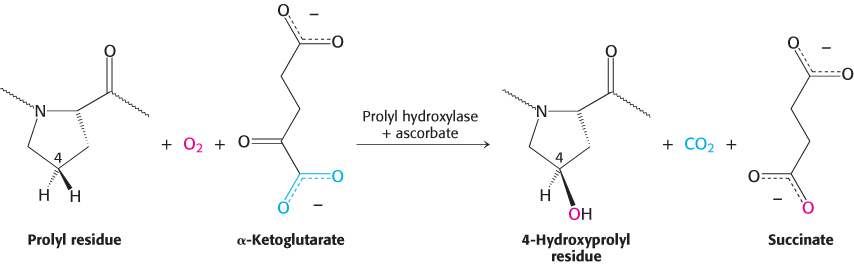
FIGURE 27.16Formation of 4-hydroxyproline. Proline is hydroxylated at C-4 by the action of prolyl hydroxylase, an enzyme that activates molecular oxygen.
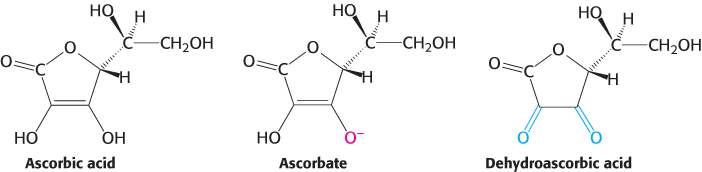
FIGURE 27.17Forms of ascorbic acid (vitamin C). Ascorbate is the ionized form of vitamin C, and dehydroascorbic acid is the oxidized form of ascorbate.