10.3 REGULATION OF CHROMOSOME STRUCTURE
Nucleosomes control the accessibility of DNA to decoding proteins such as RNA polymerase and to transcription activators and repressors. For example, nucleosome arrangements that lead to a more “open” chromatin state are associated with active gene transcription, whereas a more “closed” chromatin state typically represses transcription. The arrangement of nucleosomes on DNA is regulated by two main classes of enzymes, the activity of which usually depends on other functional subunits within a multiprotein complex.
One class of proteins that alter nucleosome arrangement is the chromatin remodeling complexes, which slide the nucleosome to a different location, eject it from the DNA, or replace it with a new nucleosome that contains a variant histone subunit (Figure 10-18a). Variant histone subunits impart special properties to the chromatin. The other class is the histone modifying enzymes, which covalently modify the N-terminal tails of histones (Figure 10-18b). Histone modifications that affect chromatin structure solely through the modification itself, without involving other molecules, are referred to as cis acting. Such a modification may result in opening or closing of the chromatin by tightening or loosening the arrangement of nucleosomes along the DNA. Histone modifications that are transacting (those that act through molecules other than the chromatin itself) attract proteins such as transcription factors or chromatin remodeling factors, which produce the chromatin change. Additional types of chemical modifications of histones are being discovered all the time.
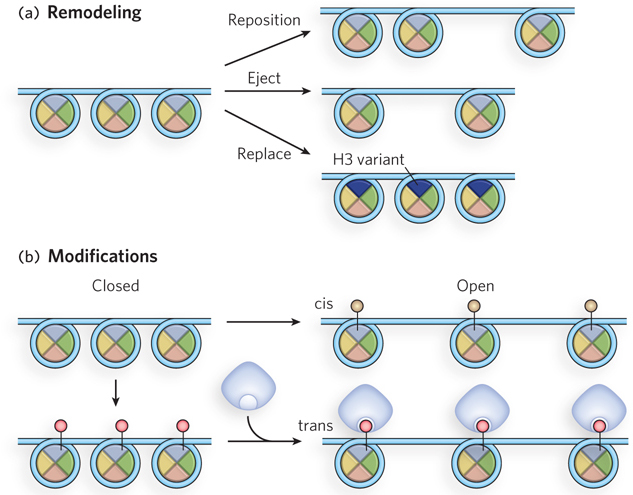
Figure 10-18: Modification of nucleosome arrangements. (a) Nucleosome position can be altered by chromatin remodeling complexes that move, eject, or replace nucleosomes. (b) Histone modifying enzymes attach chemical groups to specific amino acid residues of nucleosome subunits, as indicated by small circles attached to histones. If the outcome is a direct result of the modification, such as making the chromatin structure more open, the modification is acting in cis. If the modification attracts another protein (blue) that performs the histone-modifying function, it is acting in trans.
Enzymes that modify histones are often contained in a multiprotein complex, along with transcription activators or repressors. Nucleosome modifying enzymes and chromatin remodeling complexes can also work together. Nucleosomes are altered by the factors in specific regions of chromatin that detect environmental signals, such as transcriptional activating histone acetylases that modify histones for increased expression of particular genes. In their simplest form, nucleosome alterations control DNA accessibility by altering the structure of nucleosome-DNA complexes. DNA accessibility is essential to all types of genetic processes, including gene expression, replication, repair, and recombination.
Histone modifications are inheritable, which is especially important during an organism’s development, when a cellular program to form a specific cell type or tissue must be propagated over numerous cell divisions. Inheritance of genetic properties that are not encoded in the DNA sequence is referred to as epigenetic inheritance (further discussed later in this section).
Histone variants, along with the many covalent modifications that histones undergo, define and stabilize the chromatin state in localized regions throughout the genome. They mark the chromatin, facilitating or suppressing specific functions in a dynamic fashion, including transcription, replication, DNA repair, and chromosome segregation.
Nucleosomes Are Intrinsically Dynamic
Given the numerous contacts between nucleosomes and DNA, and the high level of DNA compaction, it seems reasonable to imagine that the condensed DNA is no longer available for other proteins to bind. However, for promoters to become accessible, the chromatin structure within the promoter region must become accessible. Molecular interactions that hold nucleosome arrays together in the 30 nm filament are likely to be relatively weak, in which case regions within nucleosome arrays will be dynamic, with hydrogen bonds between the histones and DNA breaking and re-forming quite easily.
Clever experimental designs have allowed measurements of the force on the individual molecules in chromatin. Single chromatin fibers can be isolated and chemically modified at either end, and the two ends attached to different polystyrene beads (Figure 10-19a). The bead at one end is held in a laser optical trap, and the bead at the other end is captured by a micropipette. The force required to stretch the chromatin fiber and thus disrupt the nucleosome array can be measured by moving the optical trap with a known force (measured in piconewtons; 1 piconewton (pN) = 10−12 newtons). The stretching force on a chromatin fiber produces a structural transition at just 5 pN (Figure 10-19b)—substantially less than the 10 to 15 pN needed to unzip double-stranded DNA, and much less than the 1,600 pN required to break a covalent bond. Once the chromatin fiber is stretched, the force can be relieved and the fiber then contracts to its original length, thus demonstrating that condensation of the nucleosome array is spontaneous and reversible.
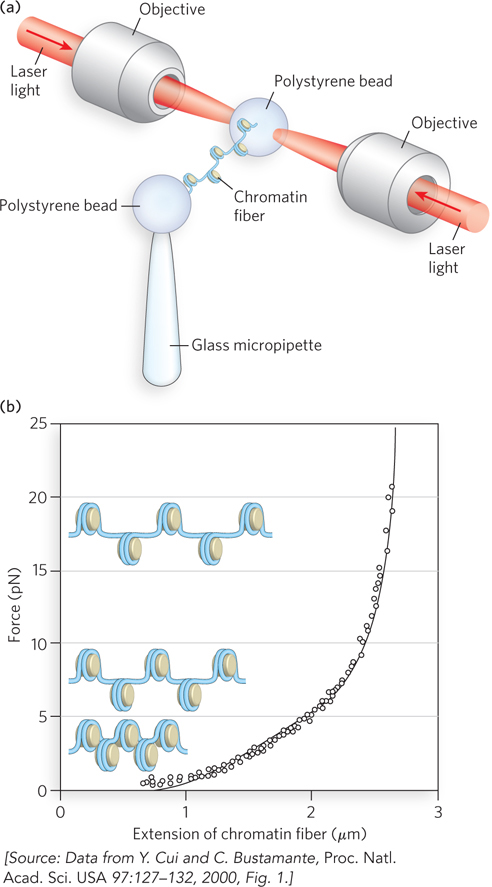
Figure 10-19: Weak internucleosomal interactions in chromatin as revealed by optical trapping. (a) The experimental setup uses DNA packaged into chromatin, with a bead coupled to each end of the DNA. One bead is held in place with a micropipette, and the other is held by an optical trap formed by a focused laser beam. The optical trap can be moved to apply a specific force on the DNA, and the length to which the DNA is stretched is measured directly. (b) Fiber length is measured (from the known magnification) at different applied forces (in piconewtons), and the process can be reversed, showing that chromatin can stretch and snap back to its original structure.
ATP-Driven Chromatin Remodeling Complexes Can Reposition Nucleosomes
Although nucleosomes can bind almost any DNA sequence, the position of nucleosomes in the genome is not entirely random. Some DNA sequences, as noted earlier, are preferentially bound by nucleosomes, but nucleosome positioning is also dictated by chromatin remodeling complexes, which use the energy of ATP to move nucleosomes around on the DNA. Chromatin remodeling complexes consist of 2 to 18 subunits and can be divided into three main classes: SWI/SNF (switch-sniff, the first remodeling complex discovered), ISWI (imitation switch), and Mi2/NURD (Table 10-2). All chromatin remodeling complexes contain a conserved ATPase subunit and use the energy from ATP hydrolysis to disrupt the many contacts between nucleosomes and DNA: this allows nucleosomes to be ejected or repositioned on the DNA. Although we lack details about the mechanisms, all chromatin remodeling complexes can be thought of as mobilizing nucleosomes on DNA. The activity of chromatin remodeling complexes can result in gene activation or repression. In general, the SWI/SNF class is associated with gene activation, and the ISWI and Mi2/NURD classes with gene repression.
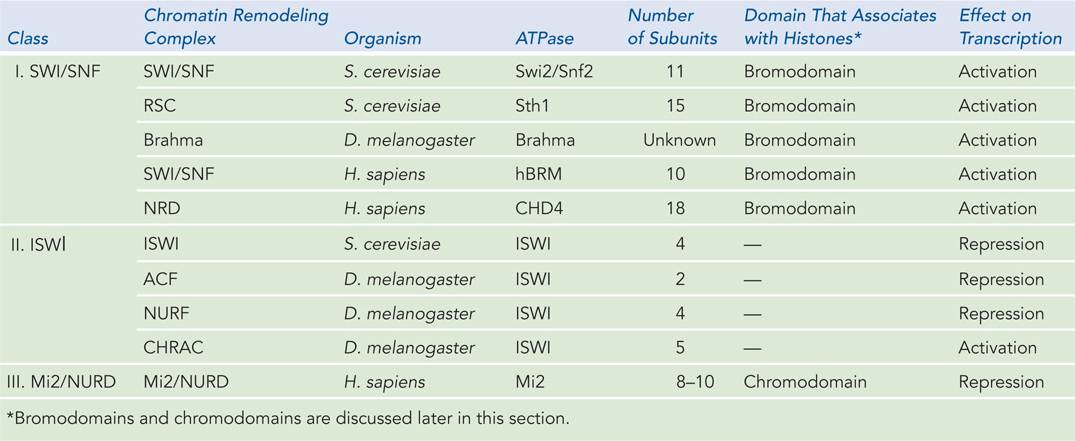
Figure 10-2: Examples of Chromatin Remodeling Complexes
The different chromatin remodeling complexes seem to have distinct mechanisms of action. Some mobilize nucleosomes by forming a DNA loop within the nucleosome (Figure 10-20a). The loop is propagated around the histone octamer, causing the nucleosome to slide to a new segment of the DNA. This can result in enhanced DNA accessibility—for example, by exposing a promoter that was previously blocked by the nucleosome. Alternatively, the same mechanism can reposition a nucleosome over a previously exposed promoter region, thereby repressing gene transcription. Likewise, repositioning nucleosomes into a regular array tends to condense the DNA, making it less accessible, whereas disrupting regularity in nucleosome arrays decondenses chromatin and thus facilitates transcription. Some chromatin remodeling complexes can also eject a histone octamer to generate a nucleosome-free region for transcriptional activation.
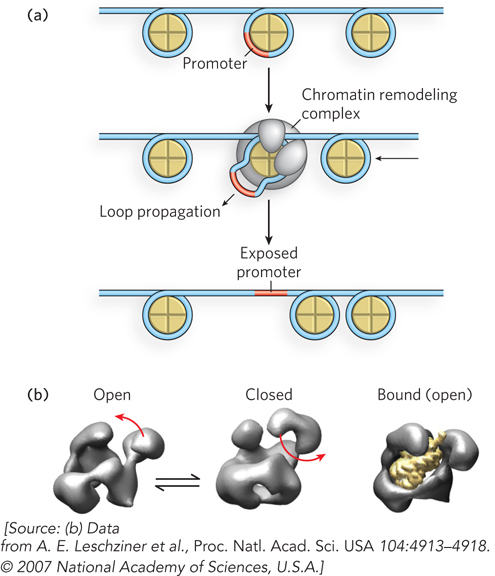
Figure 10-20: A mechanism for the action of chromatin remodeling complexes. (a) When a nucleosome covers a promoter, a chromatin remodeling complex can expose the promoter by pulling the DNA out of the nucleosome and into a loop. The loop can be propagated around the nucleosome, with the net effect of moving the nucleosome away from the promoter. (b) The shape of the chromatin remodeling complex RSC of yeast, based on three-dimensional image reconstruction from numerous images taken in the electron microscope with different views and tilts. By grouping the microscopically observed shapes into distinctive categories, different conformations can be deduced, as shown here.
The mechanistic details of how chromatin remodeling complexes function are largely unknown, and this is an exciting area of research. The low-resolution structure of a remodeling complex of yeast, for example, reveals a complex containing a jaw sufficiently wide to accommodate an entire nucleosome (Figure 10-20b).
The position of nucleosomes within a genome can be determined through the use of large-scale array technologies, such as the ChIP-Seq and ChIP-Chip techniques (Figure 10-21a). Briefly, cells are treated with formaldehyde to covalently cross-link nucleosomes to DNA. The cells are then disrupted, and genomic DNA is digested with micrococcal nuclease. An antibody to a histone is then used to immunoprecipitate the nucleosome-DNA complex. Any DNA not bound to the histone is digested and washed away, the protein-DNA cross-links are broken, and the released DNA is sequenced. This technique of chromatin immunoprecipitation followed by DNA sequencing is known as ChIP-Seq. Alternatively, the released DNA is labeled and used to probe a microarray representing the genomic sequences of that particular cell type. The pattern of hybridization on the microarray reveals the DNA sequences that associate with the nucleosomes. Because microarrays are often referred to as chips, this technique is called a ChIP-Chip experiment.
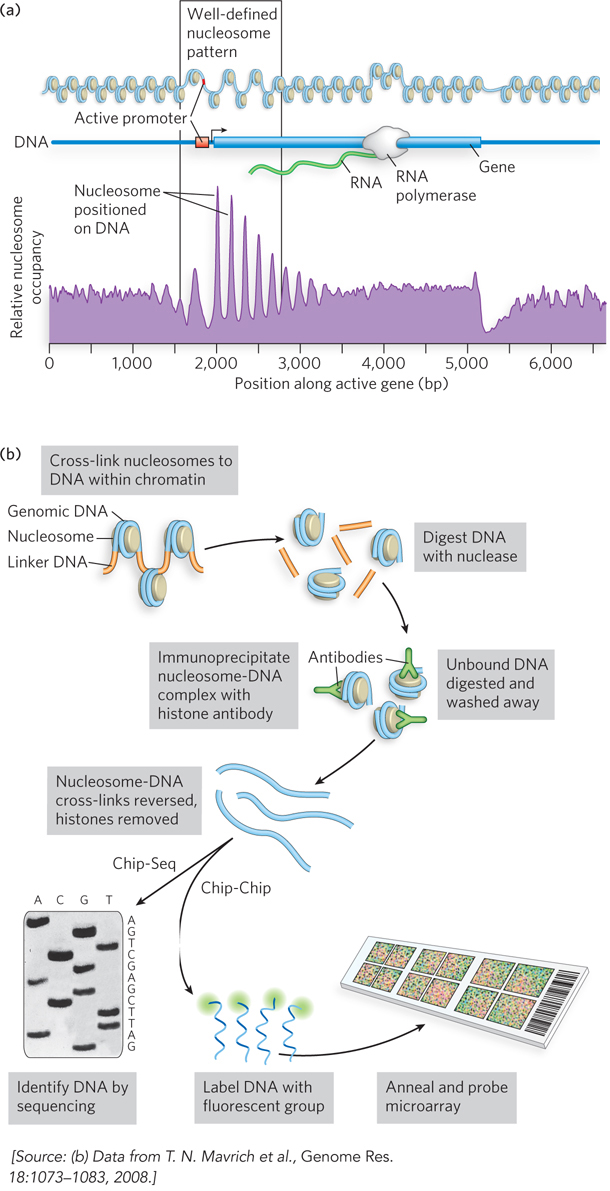
Figure 10-21: Determining the position of nucleosomes on genomic DNA with ChIP techniques. (a) Genome-wide analysis of the position of histones at all active promoter elements of yeast by the ChIP-Seq technique reveals a pattern of DNA accessibility at the promoter region. Peaks indicate regions of DNA bound to histones, and valleys indicate regions of DNA relatively free of histones. (b) Chip-Chip experiments define the location of nucleosomes positioned around an active gene.
Application of the ChIP-Seq technique has defined the position of every nucleosome in the genomes of some species of yeast, some worms, fruit flies, and humans. A consensus among this genome-wide data shows a surprisingly well-defined pattern of nucleosome position at active promoter elements (Figure 10-21b). Transcriptionally active promoters are usually free of bound nucleosomes and are flanked on either side by a nucleosome with a well-defined position. This arrangement facilitates transcription by making the promoter region accessible to transcription factors. The upstream nucleosome is the most well defined, and the DNA sequence often conforms quite well to sequences known to bind nucleosomes (see Figure 10-7), as determined by Jonathan Widom and other researchers. The position of the downstream nucleosome is also well defined; nucleosome positions farther downstream are less defined and become irregular. This pattern is observed in the genomes of all eukaryotes.
Variant Histone Subunits Alter DNA-Binding Affinity
Eukaryotes contain several variants of H2A and H3 that differ in their N- and C-terminal sequences and replace the wild-type (canonical) subunit; these changes confer special properties on the chromatin structure (Figure 10-22). The primary difference in the histone variants that replace H3 is the susceptibility of residues in the N-terminal tail to modifications such as methylation and phosphorylation. In contrast, variants of H2A differ primarily in the C-terminal tail region, which can recruit various proteins to the nucleosome. The H2A variants differ slightly in amino acid sequence, and often result in a different phosphorylation state. It is not yet clear how histone variants function, but genetic models have linked each histone variant with a specific cellular process.
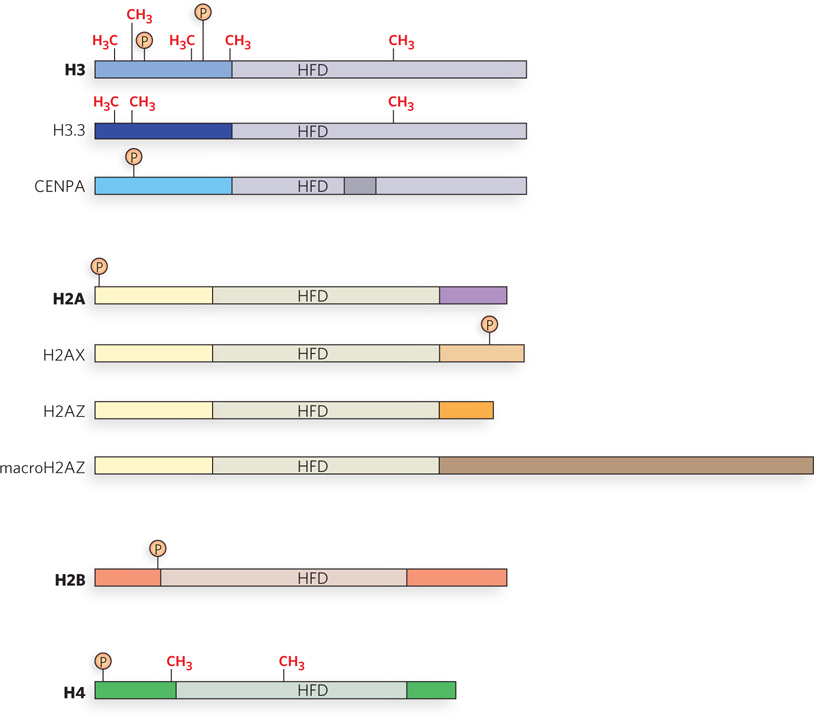
Figure 10-22: Wild-type histones and variant histones. Sites of Lys residue methylation (CH3) and Ser residue phosphorylation Ⓟ are indicated. HFD is the histone-fold domain, a structural motif common to all core histones. H3.3 and CENPA are H3 variants; H2AX, H2AZ, and macroH2A are H2A variants. The canonical (wild-type) histone subunits (H3, H2A) are shown above their variant versions. Significant regions of nonsimilarity between a canonical histone and its variant(s) are indicated by different colors. The top three illustrations show wild-type H3 and its two variants, H3.3 and CENPA; the next four show wild-type H2A and its variants, H2AX, H2AZ, and macroH2AZ. There are no known variants of H2B and H4; their wild-type versions are shown.
The wild-type H3 subunit is replaced by the H3.3 variant in regions where active gene expression is occurring (Figure 10-23a). The H2A variant H2AZ is also associated with nucleosomes located at actively transcribed genes. Based on these findings, H3.3 and H2AZ are thought to stabilize the open state of chromatin, thereby facilitating access of the transcriptional machinery to DNA in actively transcribed regions.
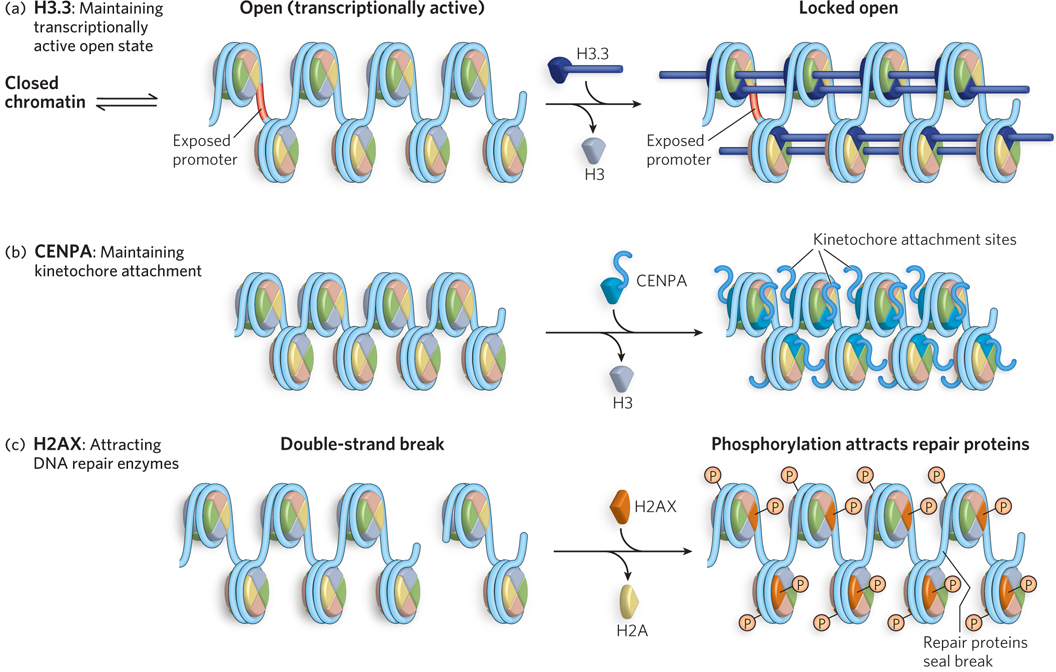
Figure 10-23: Proposed mechanisms for the roles of three histone variants. (a) The H3.3 variant is remodeled into octamers at sites of active transcription, where it helps stabilize chromatin in an open, transcriptionally active form. (b) The CENPA variant is localized to the centromere region; it contains a unique extension that may help attach the chromosome to the kinetochore. (c) H2AX localizes to sites of double-strand breaks, where it becomes phosphorylated on Ser139 and attracts DNA repair complexes to the site.
The H3 histone variant CENPA is associated with the repeated DNA sequences in centromeres and contains a large extension that connects to the kinetochore (Figure 10-23b), the site where spindle fibers attach and pull chromosomes apart during cell division (see Chapter 2). Modification of the H2A variant H2AX is associated with DNA repair and genetic recombination (Figure 10-23c). Modest amounts of H2AX seem to be scattered throughout the genome. When a double-strand break occurs, nearby molecules of H2AX become phosphorylated at Ser139 in the C-terminal region, attracting DNA repair proteins. If this phosphorylation is blocked experimentally, formation of the protein complex necessary for DNA repair is inhibited. Another H2A variant, macroH2A, is abnormally large and contains a unique C-terminal domain. MacroH2A is involved in X chromosome inactivation, a fascinating process that shuts down one of the two X chromosomes in the cells of female mammals (Highlight 10-1).
HIGHLIGHT 10-1 A CLOSER LOOK: The Use of a Histone Variant in X Chromosome Inactivation

Mary Lyon
Female mammals have two X chromosomes, and males have only one. Therefore, we might expect all the genes on the X chromosome to be expressed at twice the level in females compared with males. Most fertilized cells harboring an extra autosomal (non-sex) chromosome are lethal at the embryonic stage, due to the enhanced gene dosage from the extra chromosome. So, how do female cells, with two X chromosomes, survive with double the gene dosage? Or, how do male cells survive with half the necessary gene dosage? The answer lies in a fascinating process called X chromosome inactivation, in which one of the two X chromosomes in every cell of the female is converted into highly condensed heterochromatin, a form of very tightly compacted DNA, which silences its genes. Inactivation of the X chromosome is also known as lyonization, after its discoverer, Mary Lyon.
In the two- or four-cell stage of development of female mammals, only the paternally derived X chromosome is inactivated; gene silencing of this type, specific to either paternal or maternal genes, is known as imprinting (see Chapter 21). Both X chromosomes become active again in the early blastocyst, but one is permanently inactivated a short time later. This permanent inactivation is random; either the maternal or paternal X chromosome is targeted for inactivation. Thus, the female body is a mosaic of cells, some of which have an active maternal X chromosome and others an active paternal X chromosome. Because of the random nature of X chromosome inactivation, an animal that is heterozygous for an X-linked trait has a mosaic phenotype, as seen in the mottled coat color of a calico cat (Figure 1).

FIGURE 1 The coat color variation of a calico cat is an example of a mosaic phenotype.
The inactive X chromosome contains an abundance of the histone variant macroH2A. This variant contains a large C-terminal region that doubles the size of the protein. The current hypothesis is that the extra protein sequence binds RNA. Essential to X chromosome inactivation is the presence of a noncoding RNA called XIST (see Figure 21-20). XIST RNA is transcribed from only one X chromosome, whereupon it acts in cis to coat the entire chromosome, encapsulating it. The macroH2A histone variant binds XIST and may facilitate this process. Recent evidence suggests that macroH2A1 may be involved in maintenance of the inactive state. The condensed and inactivated X chromosome, called a Barr body, is easily identified in cells stained with an antibody specific for macroH2A1. Figure 2 shows two blastocyst cells (top and bottom) stained for total DNA (left) and the same two cells stained using an antibody to the unique C-terminal region of macroH2A1 (right), thus revealing the X-inactivated chromosome containing macroH2A1. Other modifications associated with X chromosome inactivation are cytosine methylation at CpG residues, low levels of histone acetylation, and methylation of Lys9 on histone H3. These modifications are typically associated with transcriptional repression.
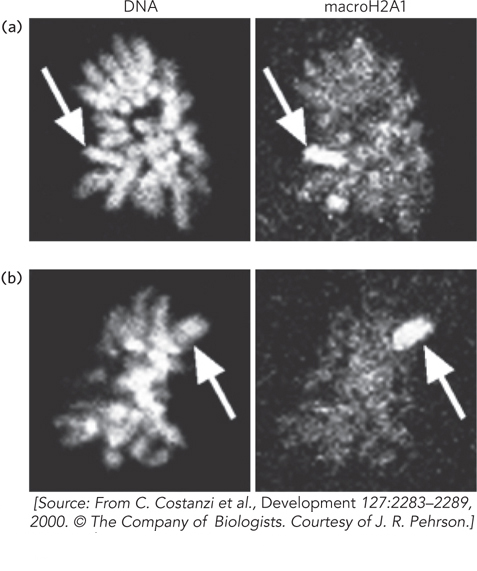
FIGURE 2 Cells of two different mouse blastocysts are stained for total DNA (left) or for macroH2A1 (right), using an antibody to macroH2A1. The arrows point to the inactive X chromosome containing macroH2A1.
Nucleosome Assembly Requires Chaperones
Isolated histones are difficult to assemble onto DNA in vitro; only low yields are obtained in the absence of other factors. In cells, histone chaperones are required to assist the assembly of histone octamers on DNA. These histone chaperones are acidic proteins that bind either the H3-H4 tetramer or the H2A-H2B dimer. Histone chaperones also function in assembling new histones on DNA during DNA replication. Chaperone-mediated nucleosome assembly occurs in two steps, as shown in Figure 10-24. First, the CAF-1 chaperone deposits an H3-H4 tetramer onto the DNA; second, the NAP-1 chaperone assembles two H2A-H2B heterodimers with the H3-H4 heterotetramer to form the complete nucleosome.
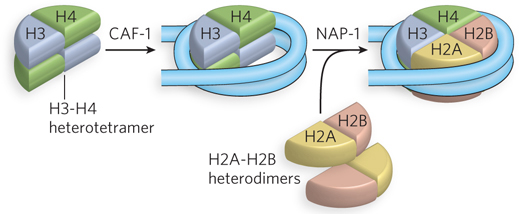
Figure 10-24: Chaperone-mediated nucleosome assembly. The H3-H4 heterotetramer is placed on DNA by the CAF-1 chaperone. Two H2A-H2B heterodimers then assemble in a reaction promoted by NAP-1.
Similar chaperones assemble the nucleosomes containing histone variants. Nucleosomes containing histone H3.3, for example, are deposited by a complex in which CAF-1 is replaced by the protein HIRA (the name is derived from a class of proteins called HIR, for histone regulation). HIRA, a transcriptional repressor, is a chromatin remodeling complex but can also be considered a histone chaperone, helping to ensure the proper assembly and placement of nucleosomes.
Modifications of Histone Tails Alter DNA Accessibility
For many years, scientists thought that histones performed only a structural role—condensing DNA. We now know that chromatin structure regulates essentially all genetic transactions and that modification of the N-terminal tails of histones plays a major role in altering chromatin structure. The amino acid residues of histone tails can be chemically modified in many ways; the most intensively studied are the acetylation of lysines, methylation of lysines and arginines, phosphorylation of serines, and ubiquitination of lysines (Figure 10-25). Figure 10-26 shows some of the most common amino acid modifications in histones.
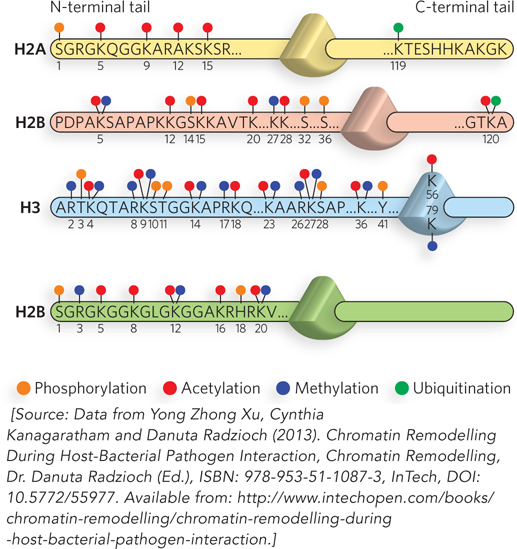
Figure 10-25: The modification of nucleosome N-terminal tails by small molecules. Modifications are shown on just one tail of each type of core histone. Residue numbers are given for the modified residues (here, shown by one-letter abbreviations and subscript numbers—S1, K5, K9, etc.). The N-terminal tail is on the left, connecting to the globular domain (indicated as a triangle), followed by the short C-terminal tail. Modifications occur at all these positions, but the most common are those in the N-terminal tails. The major modifications are represented as follows: P, phosphorylation; A, acetylation; M, methylation; and U, ubiquitination.
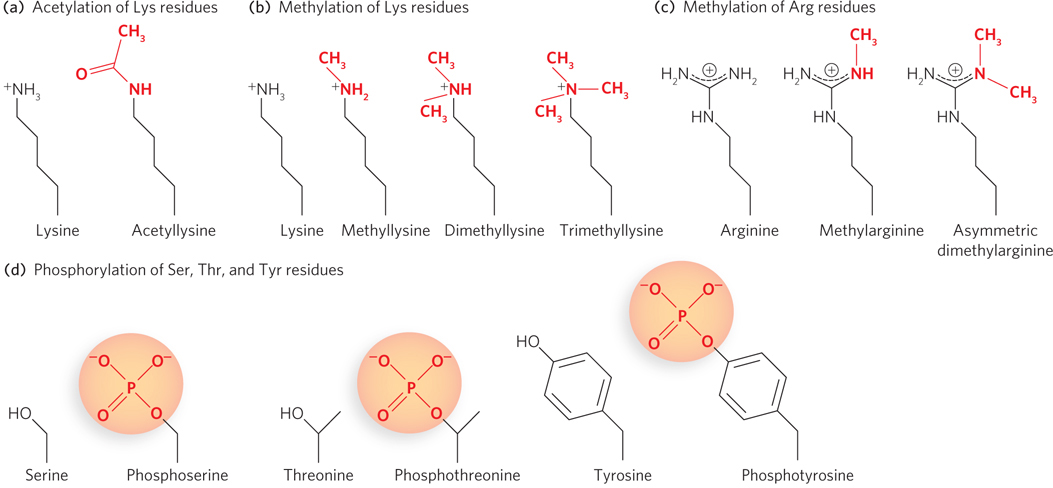
Figure 10-26: Chemical structures of some modified amino acid residues in histones. There are many types of chemical modifications to nucleosomal tails. Only the most common are shown here.
Acetylation of Lys residues, shown in Figure 10-26a, is performed by enzymes called histone acetyltransferases, or HATs (see the How We Know section at the end of this chapter). Several different types of HATs modify histone subunits, and most often they acetylate specific residues in a histone tail. Acetylation is generally associated with enhanced accessibility to DNA and subsequent transcriptional activation. Histone deacetylases (HDACs) remove acetyl groups from histones, and deacetylation of Lys residues generally results in transcriptional repression (Table 10-3). Initially, investigators thought that acetylation, which neutralizes the positive charge on lysine, simply relaxed the grip of a nucleosome on the DNA, thereby opening chromatin to regulatory proteins. More recent research indicates that acetylation has a larger effect on nucleosome-nucleosome contacts and alters higher-order chromatin structure that way, rather than changing nucleosome-DNA affinity.
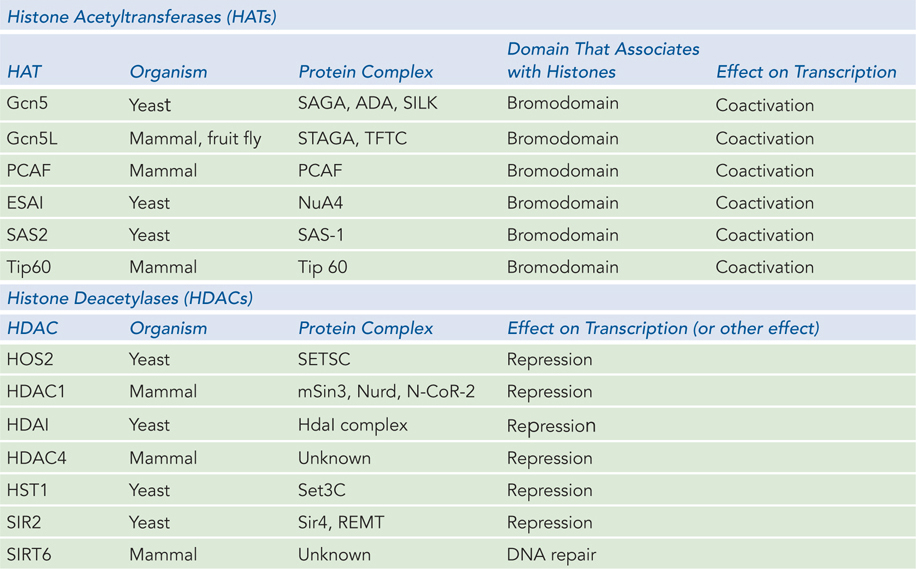
Figure 10-3: Histone Modifying Complexes
Lysine residues can also be methylated, to methyl-, dimethyl-, or trimethyllysine (Figure 10-26b). Methylation (by methylases) was once thought to be irreversible, but recent experiments have identified enzymes (histone demethylases) that remove methyl groups from histone tails. Some amino acid residues, such as Lys9 or Lys14 of H3, can be either methylated or acetylated, and this seems to stabilize the open or closed state of the nucleosome. Arginine can also be methylated, to methylarginine or two forms of dimethylarginine; double methylation can result in one methyl on each nitrogen of the guanidinium group or two methyls on one of the nitrogen atoms of the guanidinium group (Figure 10-26c).
Phosphorylation is another type of modification commonly found on histone tails of H3 and H4. The phosphorylation of Ser, Thr, and Tyr residues incorporates a negative charge into a histone tail (Figure 10-26d). As noted above, histones can also be modified by ubiquitination (not shown in Figure 10-26). Recently, histone modifications by sumoylation, proline isomerization, and ADP ribosylation have also been observed (see Chapter 21). The function of many of these modifications is still unclear.
Certain individual modifications can be correlated with specific phenotypes. While many studies are performed in yeast, our main interest is the phenotype in a multicellular organism such as ourselves. Table 10-4 provides some examples of the effects of particular histone modifications on the phenotypes of yeast and mammals.
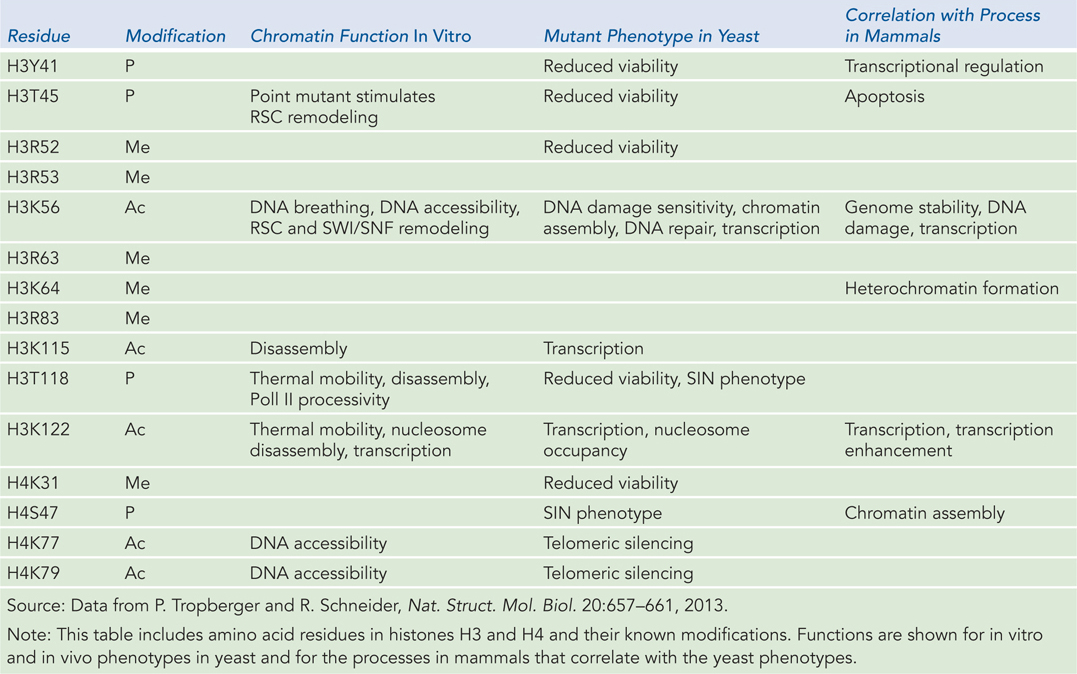
Figure 10-4: Phenotypes Associated with Modifications at Particular Residues in Histone Subunits
Proteins with Bromodomains and Chromodomains Bind Modified Histones
A more striking way in which histone tail modifications function is by recruiting enzymes that recognize particular modified amino acid residues. Proteins with bromodomain motifs recognize acetylated Lys residues (Figure 10-27). Bromodomain proteins are usually contained within a larger, multiprotein complex, such as a chromatin remodeling complex (see Table 10-2) or a complex that acetylates histones (see Table 10-3). If a chromatin remodeling complex contains a subunit with a bromodomain and also a subunit with histone acetylase activity, when the complex binds to an acetylated nucleosome it acetylates histones in a neighboring nucleosome. By this means, a specific pattern of acetylation can be propagated in a targeted area of the chromosome. Propagation of the acetylation pattern typically leads to higher levels of gene expression. Creation of a specific chromatin state underlies the epigenetic inheritance of gene expression patterns not encoded by DNA.
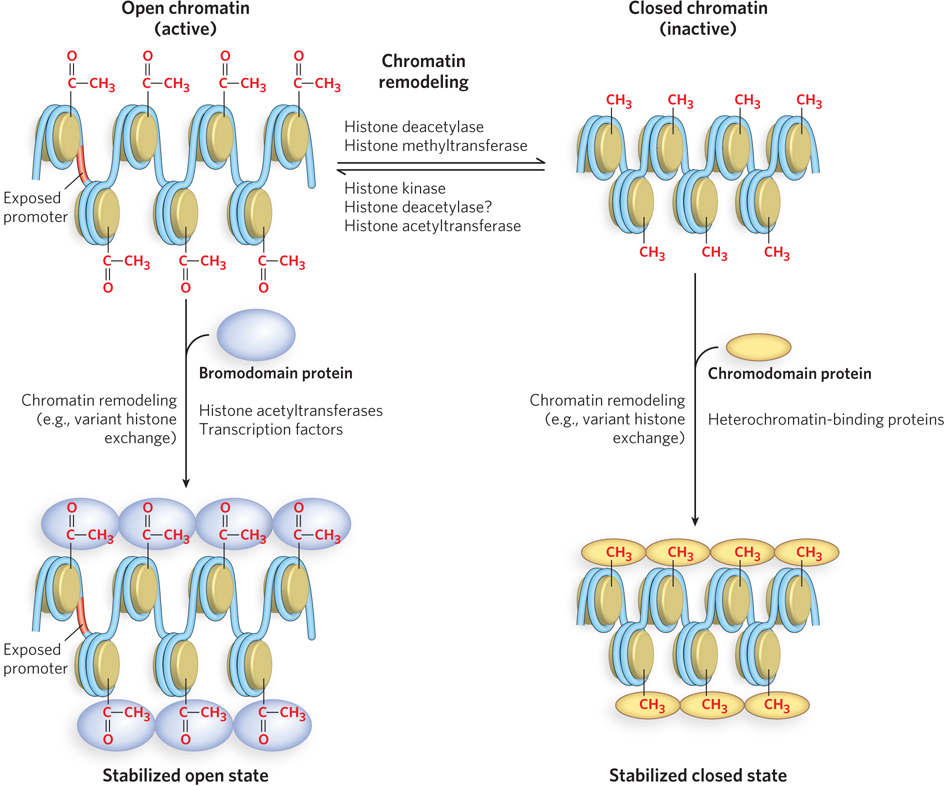
Figure 10-27: Bromodomains and chromodomains. Chromatin structure is made more open or closed by various modifications performed by the enzymes shown here. Acetylated nucleosomes (top left) are recognized by bromodomain proteins that may help stabilize the open chromatin state (bottom left). Methylated histones (top right) are recognized by chromodomain proteins that may help promote the closed state (bottom right).
As described earlier, a common modification of histone tails is methylation of Lys and Arg residues, which can result in either gene activation or gene repression. For example, in H3, methylation of Lys4 is associated with transcriptional activation, whereas methylation of Lys9 typically results in repression by recruiting chromatin remodeling complexes that condense the chromatin. Proteins that bind to methylated Lys residues contain motifs called chromodomains (see Figure 10-27). Like bromodomain proteins, chromodomain proteins can be recognized by this specific sequence motif. Chromodomain proteins are found in complexes with other enzymes that further modify chromatin structure.
Histone Modifications and Remodeling Complexes May Read a Histone Code
Some researchers have proposed that chemical modifications of histone tails can recruit proteins in a stepwise fashion, establishing a pathway that can activate the expression of a particular gene. For example, imagine a gene activation pathway that begins when a histone is acetylated. This modification recruits (via a bromodomain subunit) a HAT-containing complex that propagates the modification to neighboring histones and partially opens the chromatin structure. The resulting modifications then recruit a chromatin remodeling complex (also via a bromodomain) that completes the task of opening the chromatin structure for transcription.
The basis of the hypothetical histone code is that successive events directed by histone modifications drive transcriptional activation. A reasonable amount of research evidence supports two-step switches, in which one histone modification leads to another type of modification. Many researchers consider the histone code as still a working hypothesis, but the numerous types of histone modifications and their resulting effects provide a rich library for combinatorial regulatory signals (Figure 10-28).
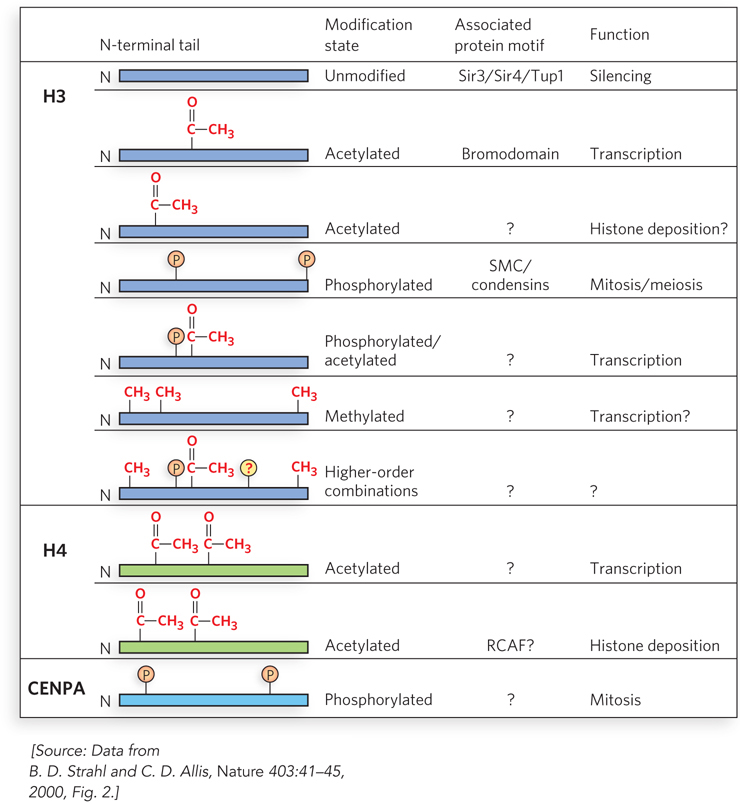
Figure 10-28: Histone modifications and their effects. Specific combinations of modifications of histones H3, H4, and CENPA (an H3 variant) are recognized by different proteins and have diverse effects on chromatin. Acetylated residues are lysines, phosphorylated residues are serines, and methylated residues are lysines and arginines.
Supporting evidence for a histone code came from studies of the expression of the human IFN-β gene (Figure 10-29). First, the Gcn5 coactivator binds within a complex of proteins to a specific DNA sequence upstream of the gene. The Gcn5 subunit is an acetyltransferase (HAT) that acetylates Lys8 of H4 and Lys9 of H3. Next, a protein kinase phosphorylates Ser10 of H3. The kinase may be a subunit of a protein complex that binds to the promoter region. This modification then promotes acetylation of Lys14 of H3 by Gcn5. These modifications, in turn, recruit a SWI/SNF chromatin remodeling complex, through a bromodomain subunit, which then remodels the chromatin and exposes a DNA sequence called the TATA box. Transcription activator TFIID binds the TATA box and, through a bromodomain, also recognizes the acetylated Lys9 and Lys14. The specific interactions of TFIID with the DNA and modified histones promote transcription. The proteins and DNA sequences involved in the initiation of transcription are discussed in greater detail in Chapter 15.
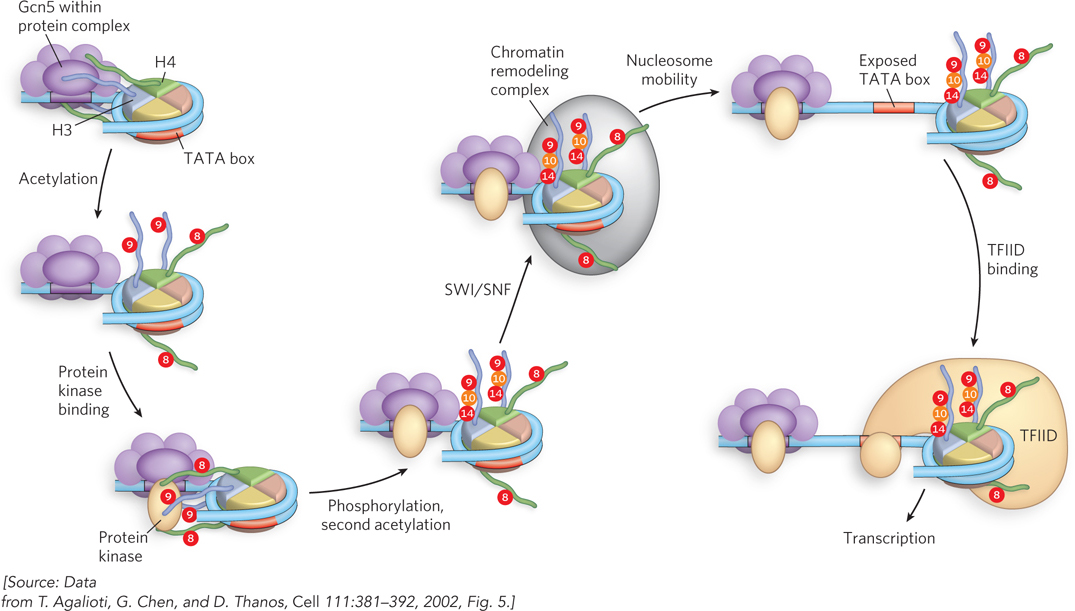
Figure 10-29: An example of a histone code. Activation of the IFN-β promoter requires an ordered succession of events driven by histone modifications that occur in a temporal sequence.
Histone Modifying Enzymes Maintain Epigenetic States through Cell Division
Certain modifications of nucleosomes are passed down to daughter cells during cell division, in mitosis and meiosis. These modifications can activate or repress certain genes. Inheriting the control of gene expression is essentially like inheriting any other trait: in addition to inheriting genes, offspring also inherit the nucleosome modifications that control expression of those genes. The underlying basis of this epigenetic inheritance is very different from classical Mendelian genetics, which is defined solely by the DNA sequence of a gene.
Histone modifications underlie the majority of epigenetic traits, although the direct methylation of cytosine bases in DNA also plays a role in epigenetic inheritance. The diversity of histone modifications provides a rich combination of modification patterns that can be coupled to epigenetic inheritance. Variant histone subunits also play a role in epigenetic inheritance in chromatin, facilitating or suppressing specific functions such as chromosome segregation, transcription, and DNA repair. Histone modifications do not disappear at cell division or during meiosis, and thus they are a part of the information transmitted from one generation to the next in all eukaryotic organisms. These modifications help define which genes are expressed by daughter cells. Epigenetic modifications are important during an organism’s development and in maintaining cell and tissue types. Epigenetic traits are also important in the process of imprinting in germ cells, in which one copy (allele) of a gene is silenced in the fertilized egg and remains turned off in every cell of the developing organism. Humans contain at least 80 known imprinted genes that are controlled by epigenetic marks, without which the fertilized egg cannot proceed past the blastula stage (see Chapter 21). The control of epigenetic marks can also be influenced by environmental factors, and when disturbed this can lead to disease, including cancer (Highlight 10-2).
HIGHLIGHT 10-2 MEDICINE: Defects in Epigenetic Maintenance Proteins Associated with Cancer
The development of cells into tissues, organs, and whole organisms requires individualized programs of gene expression for different cell types. Many of these programs are controlled by inheritable epigenetic mechanisms. Epigenetic controls often rely on chromatin structures based on specific histone tail modification patterns that provide or restrict the accessibility of DNA to transcription factors. Epigenetic controls are also maintained by chromatin remodeling complexes, histone chaperones, and DNA methyltransferases.
Molecular studies of cancer cells reveal that many types of cancer are associated with mutations in genes encoding proteins that act in epigenetic pathways. Presumably, the disruption of epigenetic pathways can activate genes that promote cell growth or repress genes that limit cell growth, leading to tumor formation. For example, aberrant histone modification patterns are observed in mammalian cancers that include mutations in certain subunits of chromatin remodeling complexes. Mutations that inactivate the SNF5 subunit, common to all SWI/SNF complexes, lead to a predisposition to familial cancer and a condition known as rhabdoid predisposition syndrome. Aberrations in certain HAT genes, as well as in particular HDACs, are associated with various hematological malignancies. Abnormal histone methyltransferases have also been linked to a predisposition to cancer. For example, loss of function of the enzyme that targets Lys9 in the N-terminal tail of H3 disrupts heterochromatin formation and could affect chromosome stability.
Epigenetic control as a basis for cancer has a positive aspect: altered epigenetic states are reversible. Cancers based in epigenetic states offer the promise of strategic therapies to block the progression of cancer cells by reestablishing the correct epigenetic state. Regaining normal gene expression by resetting epigenetic states in cell lines has already been accomplished with certain inhibitors of HATs and HDACs. These findings hold promise for future diagnostics and treatments for cancer.
Epigenetic inheritance of a chromatin state during cell division requires the preservation of histone modification patterns during DNA replication. If histone octamers were completely displaced during replication, all the epigenetic information encoded in the histone modifications would be lost in the daughter cells. Studies on the fate of individual histone subunits during replication demonstrate that histone octamers split apart during replication, but the H3-H4 tetramers remain bound to the DNA (Figure 10-30). The parental (marked) H3-H4 heterotetramers are distributed randomly on the two new daughter DNA duplexes that are made during replication, and coat only half of the total DNA after replication. New H3-H4 heterotetramers that lack the particular modification pattern of those they replace are assembled onto the replicated DNA by the CAF-1 chaperone. Current research suggests that the parental H2A-H2B dimers remain in the vicinity after being displaced by the replication fork and quickly reassemble with H3-H4 heterotetramers on the newly replicated DNA, chaperoned by NAP-1. New, unmodified H2A-H2B dimers must also be assembled on the newly replicated DNA. Hence, four types of nucleosomes form on the daughter DNA strands: nucleosomes containing (1) old (parental) H3-H4 and new H2A-H2B, (2) new H3-H4 and old H2A-H2B, (3) entirely parental H2A-H2B-H3-H4 histones, or (4) entirely new H2A-H2B-H3-H4 histones (see Figure 10-30). Overall, the newly replicated DNA has only half of the parental epigenetic information in its histones, but the daughter DNA duplexes are “salted” with the parental histone modification pattern.
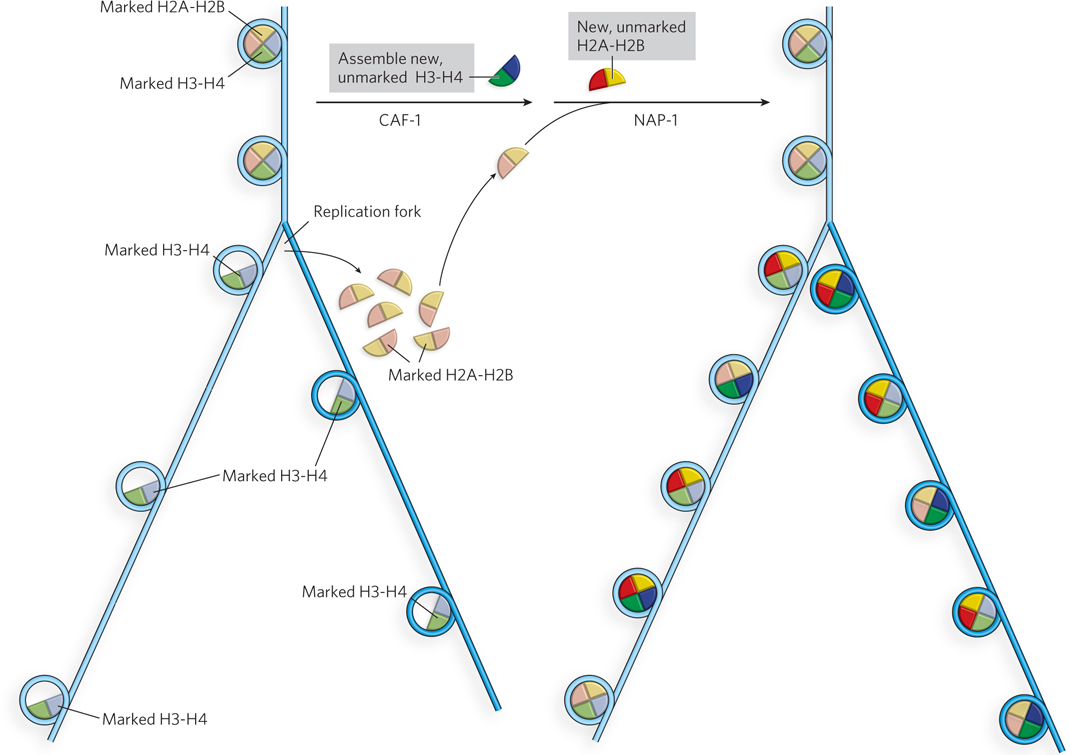
Figure 10-30: Preserving histone modification patterns during DNA replication. After DNA replication, the two new daughter duplex DNAs that are produced during replication lack histones H2A-H2B, and the H3-H4 heterotetramers distribute among the two new strands (left). The parental (marked) H3-H4 heterotetramers bind quickly to both daughter strands, along with new (unmarked) H3-H4 heterotetramers (right). The old (marked) and new (unmarked) H2A-H2B heterodimers reassemble randomly on daughter strands with old and new H3-H4 heterotetramers. The epigenetic marks are subsequently spread to adjacent nucleosomes on the two daughter strands, preserving the histone modification pattern of the parental DNA.
If half of the epigenetic histone modifications are lost during replication, how is the epigenetic state preserved in daughter cells? The answer lies in the ability of the cell to propagate the histone modification pattern of the parental nucleosomes to new histone subunits. Histone modifications are transmitted to nearby nucleosomes through the action of histone modifying complexes that recognize and bind modified residues on the parental histone subunits. For example, acetylated or methylated residues are recognized by protein complexes with specific bromodomains or chromodomains. The multiprotein histone modifying complex also contains an enzyme (e.g., HAT or methyltransferase) that spreads the modification to adjacent, unmodified nucleosomes (Figure 10-31). The different epigenetic marks are presumed to recruit specific histone modifying complexes that promote the modification of other, unmodified nucleosomes. These actions transmit and propagate the histone modification pattern to new histone subunits in the same vicinity, thereby preserving the epigenetic state of the parental cell in the two new daughter cells.
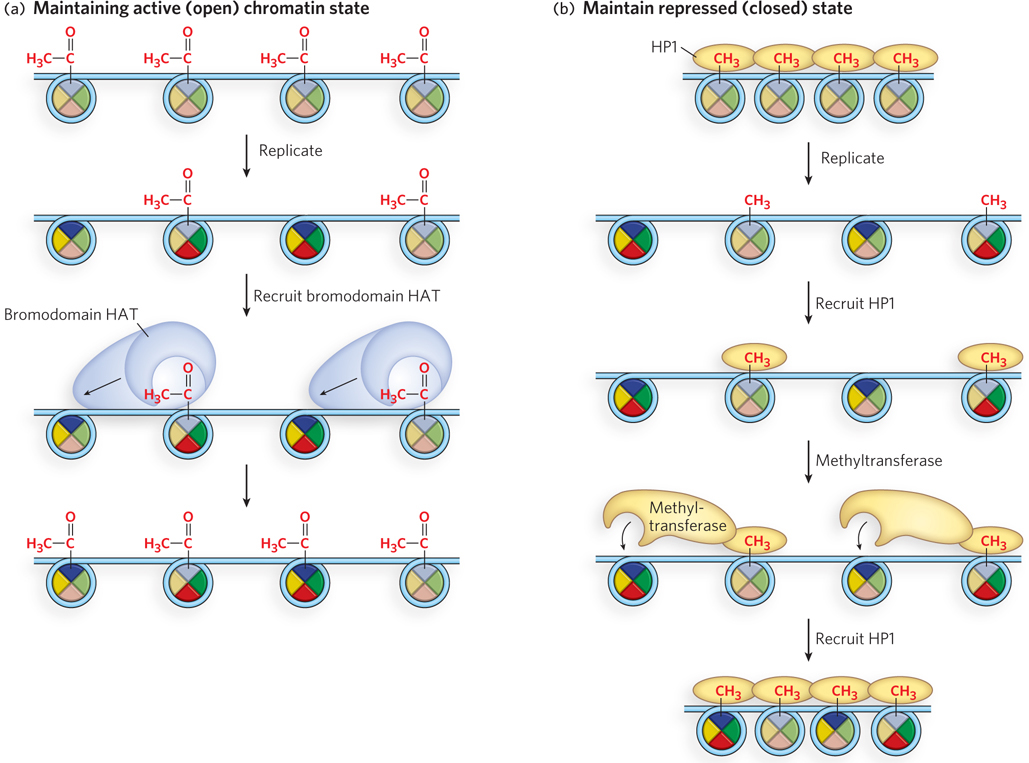
Figure 10-31: The spread of epigenetic states after replication. (a) The open state may be propagated through the binding of a bromodomain-containing complex that also contains a HAT subunit, which acetylates the same residues in neighboring histone octamers. (b) The closed state may be spread through binding of HP1, which reassociates with parental histones after replication and is associated with the repressed state of tightly packed heterochromatin. HP1 contains a chromodomain that binds methylated Lys residues in histones and the chromodomain also recruits a methyltransferase to propagate the parental methylation pattern in neighboring new histones.
SECTION 10.3 SUMMARY
Nucleosomes influence gene transcription by controlling the open or closed state of chromatin. They can be positioned at specific locations on the DNA and can slide along the duplex to allow or block access to specific regions of the DNA.
ATP-driven chromatin remodeling complexes can reposition, eject, or replace a nucleosome. The three classes of chromatin remodeling complexes are SWI/SNF, ISWI, and Mi2/NURD.
Variant histones can replace individual histone subunits in the nucleosome, thus altering the chromatin’s open or closed state.
The position of every nucleosome in a genome can be determined by ChIP-Seq and ChIP-Chip techniques.
Chemical modifications to histone tails alter the open or closed state of chromatin. Generally, acetylation is associated with enhanced accessibility of the DNA.
Proteins with bromodomains bind to acetylated Lys residues on the histone tail, recruiting other histone modifying complexes and spreading the pattern of acetylation to nearby histones. Methylated Lys residues can be bound by proteins with chromodomains, which likewise recruit histone modifying complexes that spread the methylation pattern.
The set of histone modifications in a certain region of chromatin may be read as a code that directs transcription.
Chaperone proteins are necessary for assembling histones into nucleosomes.
Epigenetic inheritance of information not encoded in the DNA relies on the transmission of histone variants and modifications through successive cell generations. Maintenance of the epigenetic state of chromatin during DNA replication requires equal distribution of H3-H4 heterotetramers on each daughter DNA. The remaining half of the parental epigenetic information is then spread to nearby newly formed histones on the two daughter strands.
UNANSWERED QUESTIONS
The organization of DNA in chromosomes is still largely a mystery. At the level of individual DNA segments, the details of how chromatin remodeling complexes and histone modifying enzymes control access to the DNA have yet to be explored. The way in which epigenetic marks are interpreted is only now coming into focus; how they are read and how they relate to normal functions and disease are poorly understood. We can expect a rapid pace of new discoveries in this exciting field of inquiry.
How is DNA packaged in a chromosome? We have an exceptional understanding of DNA condensation at the level of the nucleosome. But the arrangement of nucleosomes and their packing into the 30 nm filament account for only a 35- or 40-fold compaction of the DNA, far from the 10,000-fold compaction needed to pack the genome into the cell.
How do nucleosomes control individual gene expression? The expression of genes is controlled by many factors, including modification of the histone tails. Do individual modifications produce a series of distinct changes in chromatin, as suggested by the histone code hypothesis? If so, what are the steps that distinguish the expression of individual genes?
How are different epigenetic states specified? The exact type and combination of histone modifications that encode various epigenetic states are poorly understood, yet they are of utmost importance to human health and disease. Are epigenetic states that are inherited through generations altered by environmental factors? Can diseases that derive from an epigenetic source be reversed by therapeutic intervention? Understanding how epigenetic states are achieved and maintained is a most exciting avenue of future research.
Kornberg Wrapped His Mind around the Histone Octamer
Kornberg, R.D. 1974. Chromatin structure: A repeat in unit of histones in DNA. Science 184:868–871.
Kornberg, R.D., and J.O. Thomas. 1974. Chromatin structure: Oligomers of the histones. Science 184:865–868.

Roger Kornberg
Roger Kornberg is best known for his discovery of the structure of eukaryotic RNA polymerase II (see Chapter 15). But well before that Nobel Prize–winning work, he performed seminal studies showing that the nucleosome consists of a histone octamer with DNA wrapped around it. By the time of this work in the 1970s, scientists had already determined that the nucleosome contains four “core” histones in a 1:1:1:1 stoichiometry, and they thought that histones were arranged in some type of spiral. Kornberg’s goal was to determine the crystal structure of the histone-DNA complex. But when he purified the separate histone subunits according to established purification protocols and mixed them together, they did not assemble into an oligomer. Kornberg deduced that the pure histones failed to assemble because they were denatured by the acid and guanidine hydrochloride used in the purification.
Around this time, a new histone purification protocol was developed that did not use denaturing solvents, but the procedure went largely unnoticed because it did not separate the four histones; rather, it split them into two peaks, one containing H2A and H2B, and the other containing H3 and H4. Kornberg realized that this mild purification method probably kept the histones intact and that the two peaks might be heterodimer complexes of histone subunits that lie adjacent to each other in the nucleosome.
To test this idea, Kornberg treated the fraction containing H3 and H4 with a chemical cross-linking agent—a molecule that has two reactive groups and can covalently link two proteins, provided they are close together. In SDS–polyacrylamide gel electrophoresis (SDS-PAGE), a cross-linked H3-H4 heterodimer should produce a band that migrates at a position representing the sum of the masses of H3 and H4. Cross-linking is not 100% efficient, so the expected experimental result would be three bands, corresponding to H3, H4, and cross-linked H3-H4. Instead, Kornberg observed eight bands! The eight bands corresponded to the masses expected for an H32-H42 heterotetramer: H3, H4, H3-H4, H3-H3, H4-H4, H3-H3-H4, H3-H4-H4, and H3-H3-H4-H4 (Figure 1). Supporting this conclusion, sedimentation analysis yielded a molecular mass of 53.9 kDa for the largest H3-H4 complex, very close to the 53.2 kDa expected for a H32-H42 heterotetramer.
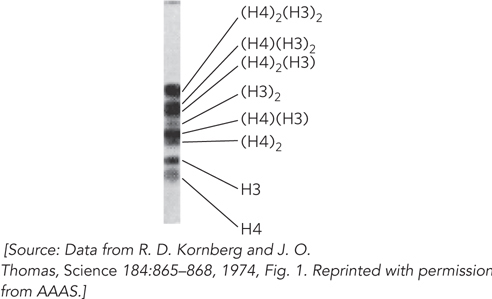
FIGURE 1 In Kornberg’s experiments, chemical cross-linking revealed that H3-H4 forms a heterotetramer. An H3-H4 complex was treated with a chemical cross-linker, followed by analysis by SDS-PAGE to separate the cross-linked proteins according to mass, which decreases from top to bottom of the gel shown here.
Combining the known 1:1:1:1 ratio of core histone subunits with his finding that H3-H4 is a heterotetramer, Kornberg hypothesized that the histone subunits assemble into an octamer containing two of each subunit. He proposed how they are arranged and surmised that about 200 bp of DNA wraps around the histone octamer, because micrococcal nuclease treatment of chromatin was known to give a repeat fragment length of 200 bp. We now know that Kornberg’s insightful conclusions were right on target!
A Transcription Factor Can Acetylate Histones
Brownell, J.E., and C.D. Allis. 1995. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc. Natl. Acad. Sci. USA 92:6364–6368.
Brownell, J.E., J. Zhou, T. Ranalli, R. Kobayashi, D.G. Edmondson, S.Y. Roth, and C.D. Allis. 1996. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84:843–851.
For many years, histones were thought to have only a structural function—packing DNA into the confines of a nucleus. We now know that histones regulate gene expression and are involved in epigenetic inheritance. Early genetic and biochemical studies of transcription factors had identified the basal RNA polymerase machinery and several transcription coactivators, but little did anyone know that many of these factors are enzymes that covalently modify histones.
With hindsight, we can see that as far back as the 1960s, researchers had correlated histone acetylation with gene regulation, suggesting that histones do not just play a structural role. The idea that histone modification could regulate transcription heated up significantly with the discovery of a nuclear histone acetyltransferase (HAT) by David Allis’s laboratory in 1995 (see this chapter’s Moment of Discovery). The Allis group chose macronuclear extracts of Tetrahymena thermophila (each cell of this organism has two nuclei: a macronucleus and a micronucleus) as an enzyme source, because this ciliate contains large amounts of acetylated histones. HAT activity was followed during purification by an activity gel assay. The assay used SDS-PAGE, starting with a gel in which histones were uniformly distributed by adding them to the polyacrylamide solution before pouring the gel and polymerizing it into a solid matrix. Column fractions of the Tetrahymena macronuclear extract were analyzed on this gel, then the gel was soaked in buffer lacking SDS, allowing proteins to renature. The buffer also contained 3H-labeled acetyl-CoA, the substrate for HAT activity. Covalent attachment of [3H]acetyl groups to the uniformly distributed histones in the gel occurred only where a HAT enzyme renatured in the gel. Unused [3H]acetyl-CoA was removed by soaking the gel, and this was followed by fluorography to visualize the position of the HAT activity, where the 3H-labeled histones overlapped with the HAT (Figure 2). The Allis lab referred to the Tetrahymena HAT as p55 because it migrates as a 55 kDa protein.
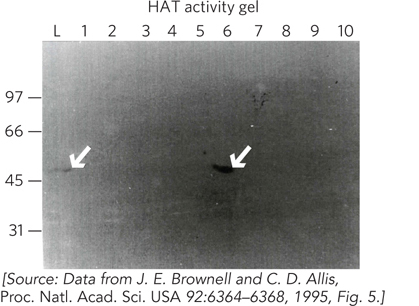
FIGURE 2 Fractions of Tetrahymena macronuclear proteins eluted from an ion-exchange chromatography column were analyzed by SDS-PAGE. Lane numbers (1 to 10) represent fraction numbers; L is the starting extract. Numbers on the left indicate molecular mass in kilodaltons. The p55 protein contains HAT activity and can be visualized in the activity gel both in the initial extract (lane L) and in the column fraction in lane 6 (arrows).
Next, the researchers identified the gene encoding p55. The sequence finding was astonishing: p55 was homologous to a very well-studied transcription activator, the yeast Gcn5 protein. This result implied that Gcn5 has HAT activity and that this enzymatic activity may account for the activator’s regulatory function. In fact, Allis’s group used their gel assay to demonstrate that Gcn5 has HAT activity (Figure 3). Since this discovery, several transcription factors have been shown to contain a subunit with HAT activity.
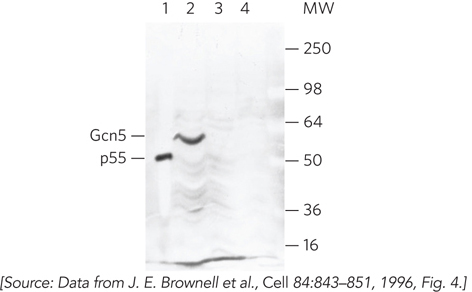
FIGURE 3 The activity gel result showing that Gcn5 has HAT activity. Macronuclear extracts of Tetrahymena show the expected HAT activity of p55 (lane 1). The gene for Gcn5 was cloned into an expression plasmid and then induced. The induced Gcn5 (60 kDa) protein has HAT activity (lane 2). Lane 3 shows the result for uninduced cells; lane 4, induced cells lacking the expression plasmid containing the Gcn5 gene. The numbers on the right indicate molecular mass in kilodaltons.