11.2 THE CHEMISTRY OF DNA POLYMERASES
The first DNA polymerase was identified in the 1950s by Arthur Kornberg and his postdoctoral fellow, Robert Lehman (see the How We Know section at the end of this chapter). In so doing, they initiated what would become an entire field of study on DNA replication enzymology. Initially, E. coli DNA polymerase I (Pol I) was simply called “DNA polymerase,” as it was presumed to be the only DNA polymerase in the cell. We now know, after decades of study, that E. coli contains five different DNA polymerases, involved in a variety of cellular processes. In fact, Pol I mainly functions in the repair of damaged DNA, although it also carries out an important function in connecting Okazaki fragments during replication, as we will see later in this chapter. We focus here on Pol I because the study of this enzyme revealed features of DNA synthesis that are common to all DNA polymerases, and it remains the most intensively studied and well-
DNA Polymerases Elongate DNA in the 5′→3′ Direction
Early work on Pol I led to the definition of two central requirements for DNA polymerization. First, all DNA polymerases require a template strand that guides the polymerization reaction according to the Watson-
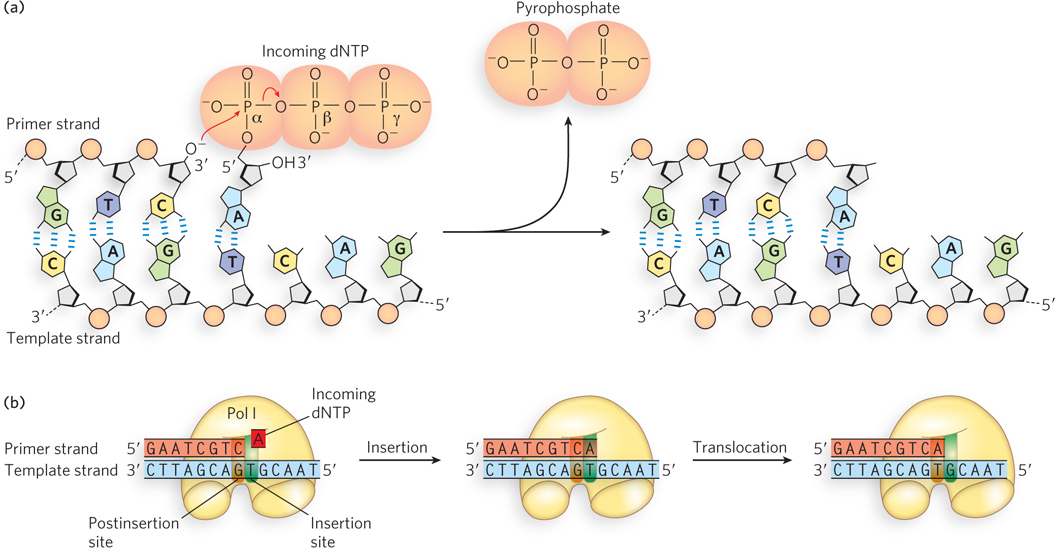
Studies of Pol I confirmed that the nucleotide precursors to DNA are the four deoxyribonucleoside 5′-triphosphates (dNTPs). The studies also showed that the different dNTPs bind the same active site on Pol I. Pol I differentiates among dNTPs only after it undergoes a conformational change that checks for the proper geometry of the base pair formed between the bound dNTP and the matching base on the template strand. Only the correct geometry of an A=T or G≡C base pair fits into the active site. An incorrect fit results in dissociation of the dNTP and binding of a new one. Normally, the polymerase is able to distinguish the correct nucleotide with this method, but one in every 104 to 105 nucleotides is added incorrectly. In the event of a misincorporated nucleotide, the polymerase has a way to remove it, which we discuss shortly.
370
The 3′-hydroxyl group of the primer strand is activated to attack the α phosphorus of the incoming dNTP, resulting in attachment of a dNMP to the primer 3′ terminus and release of pyrophosphate (PPi) (see Figure 11-5a). The overall reaction—
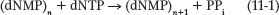
Following incorporation of a dNMP, Pol I must slide forward on the new 3′ terminus to incorporate another dNTP. Therefore, the DNA polymerase active site must be divided into at least two distinct sites (Figure 11-5b). The template nucleotide that will pair with the incoming dNTP is positioned in the insertion site. The primer strand 3′-terminal base pair is positioned in the postinsertion site. The 3′ OH on the terminal ribose of the primer strand then attacks the phosphodiester bond that connects the α and β phosphates of the incoming dNTP. This results in addition of one dNMP to the primer strand and the release of pyrophosphate. After addition of a dNMP to the primer terminus, the new terminal base pair occupies the insertion site and must be translocated to the postinsertion site, allowing the next template nucleotide and a new dNTP to occupy the insertion site. Translocation of DNA can occur by sliding of the enzyme or dissociation of the enzyme from the DNA, followed by rebinding with the terminal base pair in the postinsertion site.
DNA synthesis proceeds with only a minimal change in free energy, given that one phosphodiester bond is formed (with the addition of one dNMP to the 3′ primer terminus) at the expense of another, similar bond (between the α and β phosphates of the dNTP). However, noncovalent base-
371
At first glance, the use of a dNTP to form one dNMP link to DNA might seem to be a waste of energy. Why not use dNDP as the nucleotide precursor, which would produce the same DNA product and one inorganic phosphate (Pi) instead of PPi, which is then split into two Pi? The drawback would be that the reverse reaction could easily be initiated at any time, because Pi, the molecule that would initiate the reverse reaction, is abundant in the cell. Using triphosphate precursors ensures that the reverse reaction will not occur, because, as described above, PPi is eliminated by pyrophosphatase—
The initial studies of Pol I, performed decades ago, demonstrated that it requires a primed template and dNTP precursors as substrates. Many different DNA polymerases have been studied over the years, from sources as diverse as phages and other viruses, various types of bacteria, archaea, and eukaryotes, and mitochondria from many different species. All known DNA polymerases use the mechanism shown in Figure 11-5. Indeed, RNA polymerases also use this same basic mechanism (described in Chapter 15).
Most DNA Polymerases Have DNA Exonuclease Activity
DNA nucleases are a class of enzymes that degrade DNA. Nucleases that shorten DNA from the ends are called exonucleases; endonucleases cut DNA at internal positions. All cells have several nucleases of both types that are used in various tasks. A biochemist who wants to study any other type of enzyme that uses DNA as a substrate must carefully purify the enzyme and remove all nucleases, otherwise the DNA will be destroyed by the nuclease activity, and the researcher will be unable to observe other enzymatic reactions that require DNA. This careful purification is what Kornberg thought he was doing as he purified Pol I from a cell extract, but he was troubled by his inability to separate what he thought was a contaminating exonuclease activity from Pol I. He finally had no choice but to come to the paradoxical conclusion that the same enzyme that makes DNA also degrades it. In fact, Kornberg found that Pol I has two different exonuclease activities: one starts at the 3′ end and degrades DNA in the 3′→5′ direction (opposite to the direction of DNA synthesis), and the other starts at the 5′ end and degrades DNA in the 5′→3′ direction. The active sites of the two DNA exonucleases of Pol I are distinct from each other and from the DNA polymerase active site.
KEY CONVENTION
Exonucleases that digest a DNA strand from the 3′ terminus are called 3′→5′ exonucleases, because the strand shortens at the 3′ end while the 5′ end remains intact. In contrast, 5′→3′ exonucleases digest DNA from the 5′ terminus, while the 3′ end remains intact.
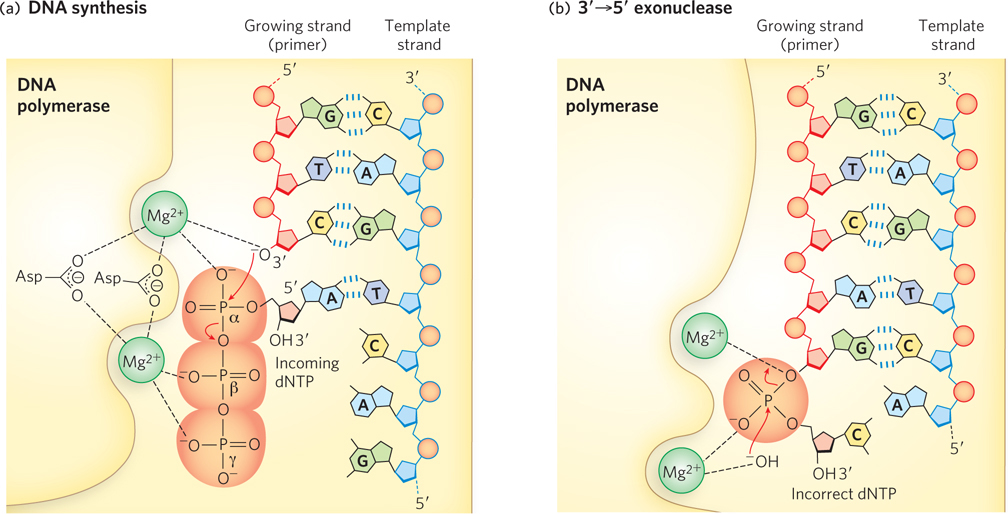
The 3′→5′ Exonuclease As noted previously, DNA polymerases are typically very accurate and produce only about one error every 104 to 105 nucleotides incorporated, by incorrect base selection. This error rate is improved 102- to 103-fold by a polymerase’s 3′→5′ exonuclease activity. When an incorrect dNMP is incorporated, the 3′→5′ exonuclease removes the mismatched nucleotide, giving the polymerase a second chance at incorporating the correct one (Figure 11-6). This activity, known as proofreading, is not the same thing as pyrophosphorolysis, the reverse of the polymerization reaction—
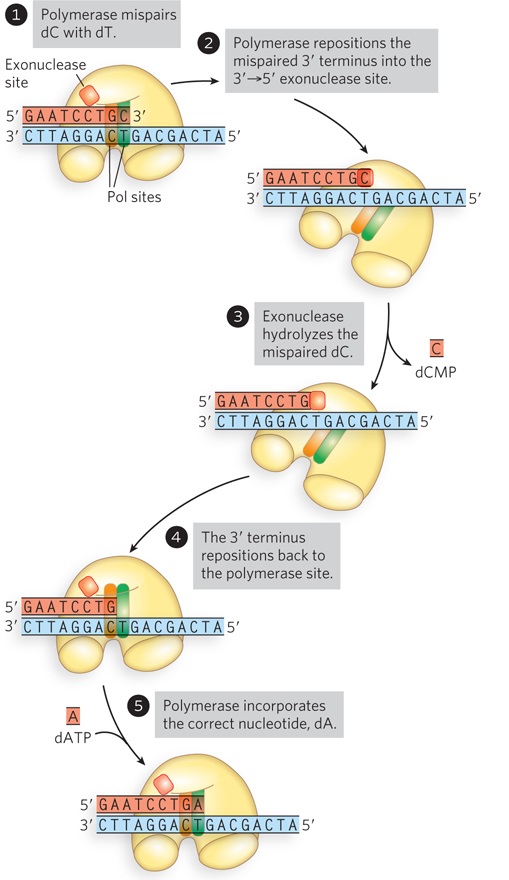
372
One way in which DNA polymerase makes errors is by incorporating dNTP tautomers (see Chapter 6) at the polymerase active site. Purine and pyrimidine tautomers can form non–
When base selection and proofreading are combined, Pol I leaves behind one net error for every 106 to 108 nucleotide additions. The DNA polymerase involved in chromosome replication has a similar error rate. How accurate must a DNA polymerase be to replicate the E. coli genome without making a mistake? Replication of the 4.6 × 106 bp (4.6 Mbp) chromosome requires polymerization of 9.2 × 106 dNTPs. An error rate of about 1 in 107 would result in only one incorrect nucleotide insertion per cell division. In fact, the observed accuracy of the overall replication process in E. coli is one error in 109 to 1010 polymerization events. The additional accuracy derives from a repair system that recognizes and removes mismatches that escape both the polymerase and the proofreading exonuclease activities (see Chapter 12). At this level of accuracy, only a single error is acquired in every 100 to 1,000 new cells.
The 5′→3′ Exonuclease Pol I also has a second exonuclease that degrades DNA in the 5′→3′ direction, the same direction as DNA synthesis. The 5′→3′ exonuclease is unique to Pol I and reflects the enzyme’s role in DNA repair. Pol I performs a host of clean-
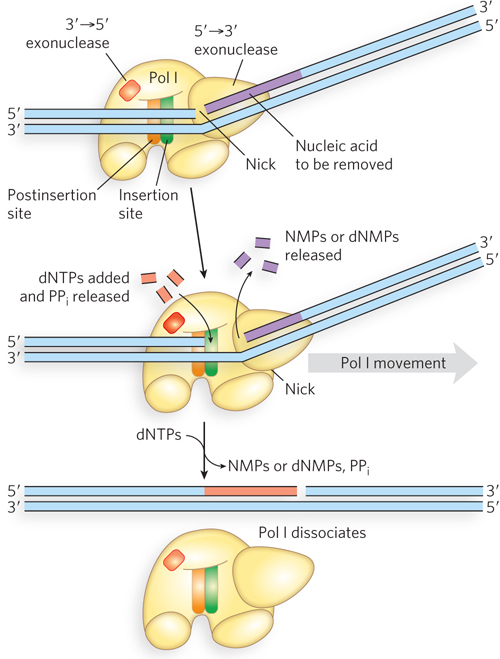
373
Five E. coli DNA Polymerases Function in DNA Replication and Repair
Escherichia coli contains five different DNA polymerases (Table 11-1). The large excess of intracellular Pol I delayed the discovery of the other DNA polymerases. Then, in the 1970s, DNA polymerase II (Pol II) and DNA polymerase III (Pol III) were discovered in studies using a mutant strain of E. coli called polA, which is depleted of most Pol I. These studies showed that Pol III is the DNA polymerase that replicates the chromosome; it is sometimes referred to as a replicase, or chromosomal replicase. Pol II seems to be involved in DNA repair.
Pol IV and Pol V were not discovered until 1999. They are different from the other DNA polymerases in that they lack a 3′→5′ proofreading exonuclease and thus often incorporate the wrong nucleotide. These low-
DNA Polymerase Structure Reveals the Basis for Its Accuracy
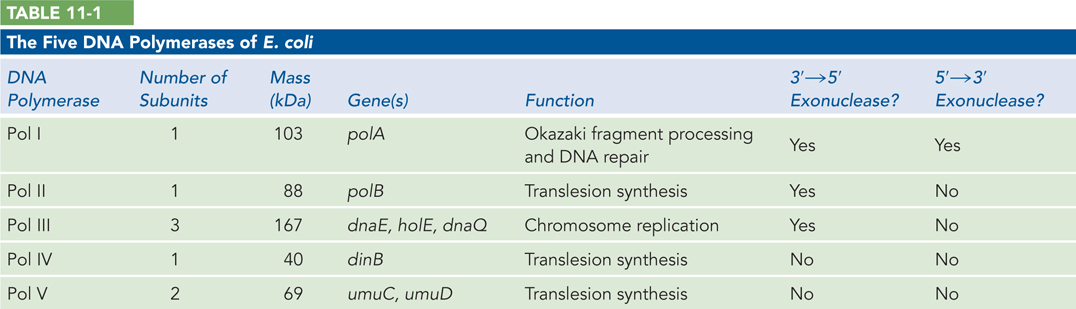
The crystal structure of E. coli Pol I resembles a right hand, with domains referred to as the palm, thumb, and fingers. All DNA polymerases have these same structural features. The bound DNA lies on the palm domain, which contains the polymerase active site and is the most conserved feature among all DNA polymerases. The fingers domain contains the dNTP-
374
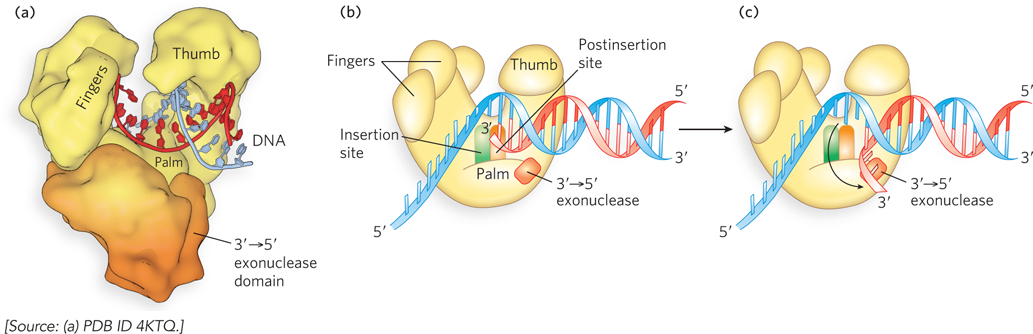
A dramatic conformational change occurs on the binding of a correct dNTP to the fingers domain. The domain rotates inward about 40°, carrying the dNTP down to the DNA template. In so doing, the enzyme forms an active-
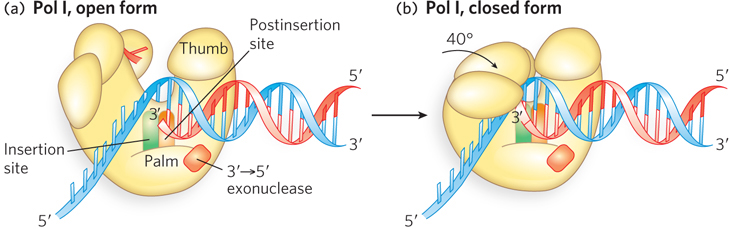
375
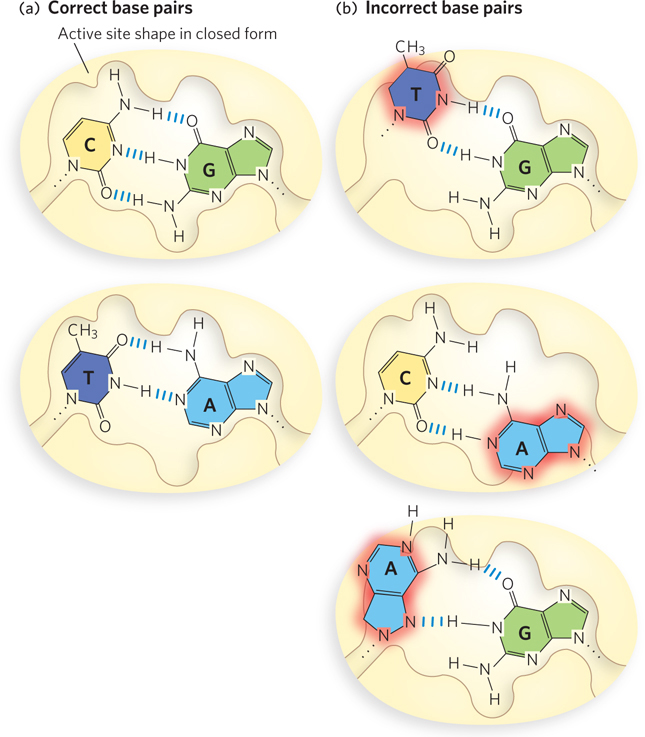
Incorporation of an incorrect base pair is 10-
 When an incorrect dNTP enters the insertion site on Pol I, binding is readily reversed (back arrow).
When an incorrect dNTP enters the insertion site on Pol I, binding is readily reversed (back arrow).  In the rare instance of incorporation of the incorrect dNTP, the process is slow due to the imperfect active-
In the rare instance of incorporation of the incorrect dNTP, the process is slow due to the imperfect active-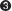 In the favored (rapid) route, the mispaired 3′ terminus is shifted to the 3′→5′ exonuclease, and in
In the favored (rapid) route, the mispaired 3′ terminus is shifted to the 3′→5′ exonuclease, and in  the mismatched nucleotide is excised to re-
the mismatched nucleotide is excised to re- This allows Pol I to insert the correct nucleotide on the second try. Binding and incorporation of a correct dNTP is rapid and paves the way for the next round of incorporation.
This allows Pol I to insert the correct nucleotide on the second try. Binding and incorporation of a correct dNTP is rapid and paves the way for the next round of incorporation. 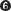 If the incorrect nucleotide remains, the mismatched DNA is slow to act as substrate for the next round of dNTP incorporation.
If the incorrect nucleotide remains, the mismatched DNA is slow to act as substrate for the next round of dNTP incorporation.The polymerase active site contains two magnesium ions that are held in place by conserved Asp residues (Figure 11-12a). One Mg2+ deprotonates the primer 3′-OH group to form the 3′-O− nucleophile. The other binds the incoming dNTP and facilitates departure of the pyrophosphate leaving group. This two-
Surprisingly, certain mutations result in DNA polymerases that are even more accurate than the wild-
376
Processivity Increases the Efficiency of DNA Polymerase Activity
During DNA synthesis, the DNA product must be moved to the postinsertion site so that the new 3′ terminus lies in the insertion site for addition of the next dNTP. To accomplish this repositioning, a polymerase can take either of two paths. It can fully dissociate from the DNA and rebind the primer strand terminus in the postinsertion site; this dissociation followed by rebinding at each nucleotide addition is referred to as distributive synthesis. Alternatively, the polymerase may simply slide forward one base pair along the DNA to reposition the 3′ terminus, without dissociating from the DNA. When a polymerase remains attached to DNA during multiple catalytic cycles, the process is known as processive synthesis.
The “processivity number” is the average number of nucleotides incorporated before the enzyme dissociates from the DNA. Processivity can result in exceedingly efficient polymerization, because much time is wasted by a dissociated polymerase in locating and rebinding a 3′ primer strand terminus. Pol I has a processivity number of 10 to 100 nucleotides, depending on conditions. In contrast, Pol III, like most DNA polymerases that replicate chromosomes (i.e., replicases), has a processivity number in the thousands, which, as we will see, results from a protein ring that encircles the DNA.
SECTION 11.2 SUMMARY
DNA polymerases require a primed template and extend the 3′ terminus of the primer strand by reaction with dNTPs.
A dNTP that correctly base-
pairs to the template strand is incorporated into the primer strand with the release of pyrophosphate. Pyrophosphate is hydrolyzed by pyrophosphatase, which reduces the concentration of pyrophosphate in the cell and makes the reverse reaction extremely unlikely. DNA polymerases are inherently very accurate and are made even more accurate by a proofreading 3′→5′ exonuclease.
Pol I also has a 5′→3′ exonuclease that degrades DNA while the polymerase synthesizes DNA, in the process of nick translation.
E. coli has five DNA polymerases. Pol III is responsible for chromosome replication, and Pol I is used to remove RNA primers and fill in the resulting gaps with DNA. The other three (II, IV, and V) are involved in DNA repair and in moving replication forks past sites of DNA damage.
Binding of a dNTP to a DNA polymerase results in a large conformational change, yielding an active site in which only correct base pairs fit.
DNA polymerases often have high processivity, in which many nucleotides are added to a DNA chain in one polymerase-
binding event.
377