11.5 TERMINATION OF DNA REPLICATION
In both bacteria and eukaryotes, replication forks meet head-
E. coli Chromosome Replication Terminates Opposite the Origin
In E. coli, a region located halfway around the chromosome from oriC contains two clusters of 23 bp sequences called Ter sites (termination sites; Figure 11-33). The two clusters of Ter sites are oriented in opposite directions. The monomeric Tus protein (termination utilization substance) binds tightly to a Ter site and blocks the advance of the replication fork by stopping DnaB helicase. A fascinating property of the Tus-
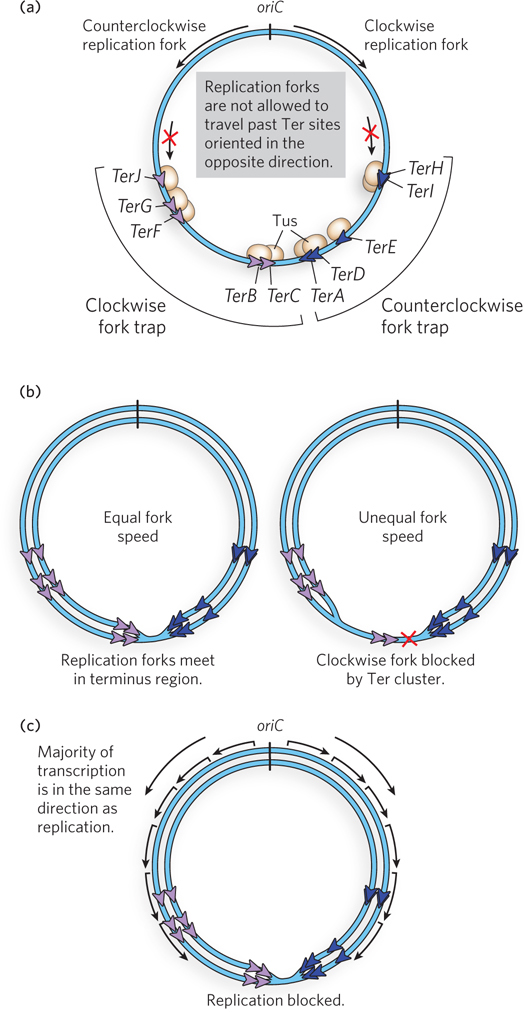
The arrangement and orientation of Ter sites is such that bidirectional replication forks from oriC can pass through the first set of Ter sites that they encounter, but are blocked by the second set. This arrangement localizes the replication fork collision zone to the area between the two clusters of Ter sites. Although Tus is not essential for E. coli growth, the Tus-
Actively replicating bacteria are also growing and metabolizing, and therefore are actively transcribing RNA from promoters throughout the chromosome. This means that collisions between RNA polymerase and replication forks are inevitable. In vivo studies show that codirectional collisions do not impede forks, whereas head-
399
Replicating the last bit of DNA between converging replication forks presents certain topological problems that must be solved to disentangle the two daughter chromosomes. Specialized type II topoisomerases unlink the catenated daughter chromosomes (see Chapter 9).
Telomeres and Telomerase Solve the End Replication Problem in Eukaryotes
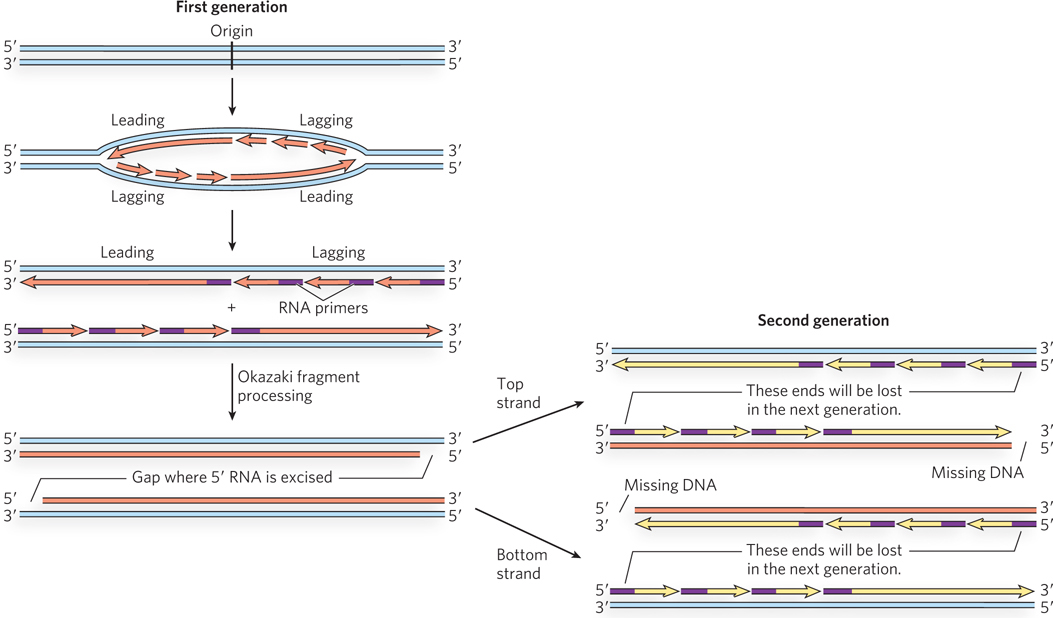
The replication of linear chromosome ends poses a unique problem. At the end of a chromosome, after the leading strand has been completely extended to the last nucleotide, the lagging strand has a single-
The ends of the linear eukaryotic chromosomes are called telomeres (see Chapter 12). How were telomeres discovered, and what is so special about them? This story starts in the 1930s with Barbara McClintock’s research on maize. McClintock noticed that when chromosomes become damaged and break in two, the broken chromosomes can rapidly “heal” by joining the two chromosome ends. When parts of two different chromosomes become joined, it sometimes creates an unusual chromosome with two centromeres. On entering cell division, chromosomes with two centromeres are pulled to opposite poles by the mitotic spindles, breaking the chromosomes, and this is followed by another round of healing. The process of healing and breaking occurs over and over as cells divide, leading to chromosome rearrangements, loss of genetic information, and cell death. It would, of course, be disastrous if the ends (telomeres) of normal chromosomes underwent this “healing” process, because all the chromosomes would break on entering cell division. Therefore, McClintock surmised, there must be something special about the ends of normal chromosomes, preventing them from being joined together by the healing process that mends broken chromosomes. Independently, Hermann Mueller, working with irradiated flies, made similar observations on the fate of broken chromosomes (in this case, broken by x rays) and hypothesized that the ends of chromosomes have a protective quality about them. He called the ends “telomeres,” derived from the Greek telos (“end”) and meros (“part”).
400


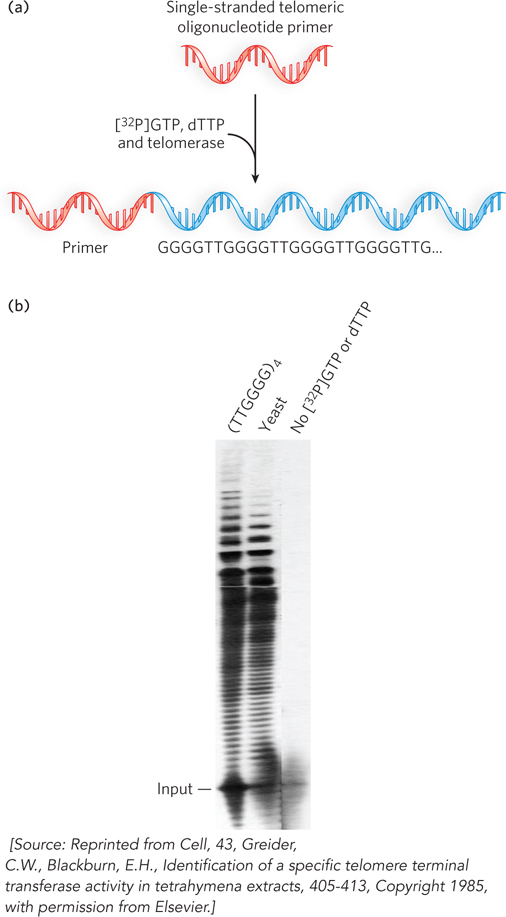
In 1978, Elizabeth Blackburn identified the first telomere sequence in the single-
The finding that yeast telomere sequences were added to the Tetrahymena telomere sequence suggested that telomeres may be synthesized by a polymerase that can synthesize DNA from an exogenous template, presumably an RNA molecule. DNA polymerases that use RNA as a template were first found in certain viruses and are called reverse transcriptases, because they transcribe RNA into DNA—
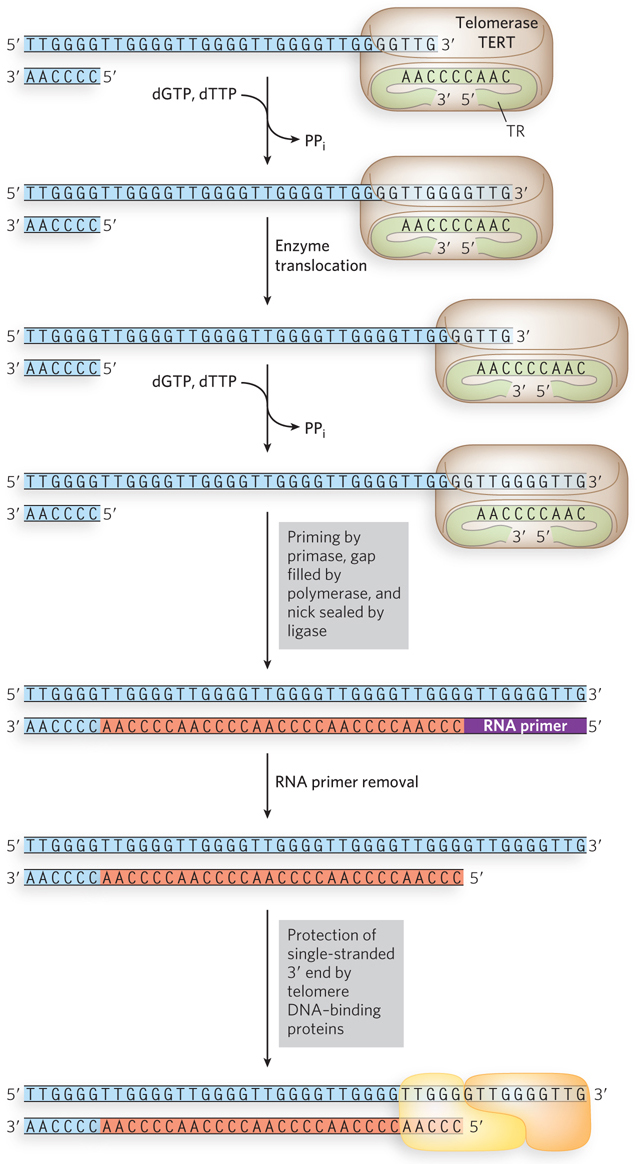
After purification and characterization, the telomerase reverse transcriptase (TERT) was found to carry a tightly bound, noncoding telomerase RNA (TR). The TR in humans is 451 nucleotides long, but can range from 150 to 1,300 nucleotides, depending on the organism. The TR contains about 1.5 telomere repeat units that it uses as a template to extend the 3′ terminus of the telomere (Figure 11-36). The TERT-
401
Telomere Length Is Associated with Immortality and Cancer
Tetrahymena, like other single-
The fact stands that telomerase is associated with cancer. HeLa cells are an immortal human cell line derived from a woman named Henrietta Lacks, who died of ovarian cancer in 1951. HeLa cells express telomerase and have been grown in cell culture for decades in laboratories around the world. If telomerase could be inhibited by a drug, the telomeres in cancer cells should shorten with each cell division until the telomeres collapse, causing cell death. Therefore, drugs that inhibit telomerase hold promise as an anticancer therapy, an active area of current research.
Telomeres are Protected and Regulated by Proteins
The linear ends of eukaryotic chromosomes present another problem: they could be mistaken for sites of chromosome breakage and thereby induce the cell’s DNA repair systems that would join the ends of chromosomes together. The cell has two main repair systems that use recombination to join broken DNA ends (see Chapter 13). This would have disastrous consequences because, as described earlier, the joined chromosomes would be torn apart during cell division. In the cell, telomeric ends are protected by specialized telomere DNA–
402
403
HIGHLIGHT 11-2 MEDICINE: Short Telomeres Portend Aging Diseases
Loss of chromosome ends during successive cell divisions leads to cell senescence or apoptosis, whereas expression of telomerase imparts immortality to cells. Hence, we may think of telomeres as a type of molecular clock that counts down the age of our cells. Can immortality be achieved by activating telomerase activity? Unfortunately, activation of telomerase in every cell of the body is not an option. Activation of telomerase is associated with cancer, and, in fact, mice that are engineered to express telomerase in somatic cells develop tumors and die early. We are caught in a delicate balance between requiring telomeres for life but also requiring that they have a finite lifetime. Given the connection between telomeres and cancer, one possible explanation for the telomere clock is that telomere shortening may have evolved as a way to suppress tumor growth in multicellular organisms. An equally feasible explanation is that natural selection favors a finite lifespan, because it ensures diversity in the genetic pool for evolution.
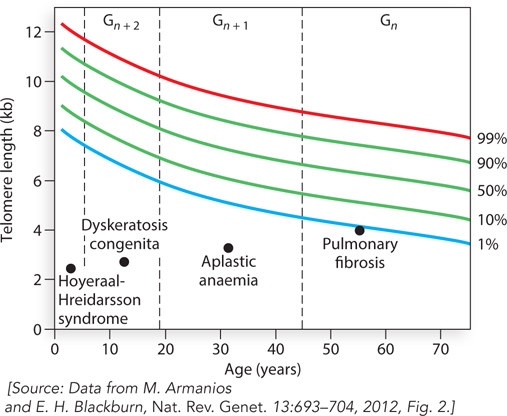
Does telomere length correlate with longevity? Several observations indicate a role of telomeres in cellular aging. A large body of epidemiological studies on human telomeres in blood cells reveals that short telomeres correlate with a number of diseases related to aging. Abnormally short telomeres are associated with diabetes, osteoporosis, obesity, cancers, impaired function of the immune system, a variety of cardiovascular diseases, and stroke. Thus, short telomeres correlate with a broad general syndrome of diseases that reflect, or perhaps even cause, aging in a fundamental way. In sum, long telomeres may impart not necessarily a longer lifespan but rather a healthier life for people in their seventies. Telomere-
Some hereditary human diseases have their basis in telomerase or in proteins that bind telomeres. Such mutations are present in individuals with dysfunctional telomeres and particular types of degenerative diseases. Importantly, these degenerative diseases are age-
Telomere-
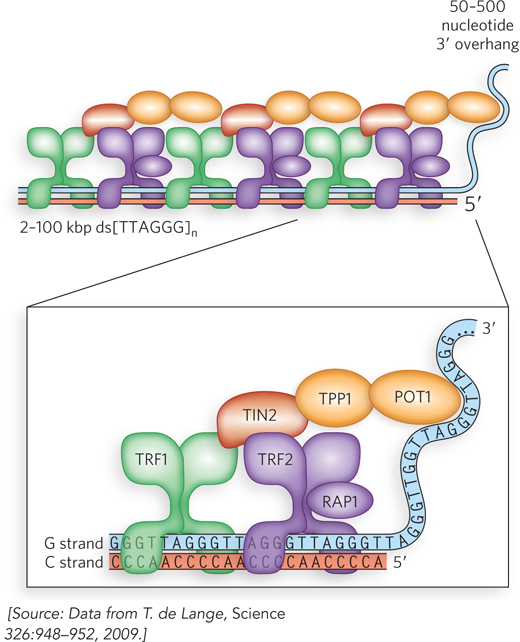
Shelterin not only protects telomeres against chromosome joining but also regulates telomere length. POT1 inhibits telomerase and becomes a stronger inhibitor with each additional POT1 protein bound to the single-

404

Telomere DNA–
In contrast to the broad conservation of telomerase among different species, the proteins that bind telomeres often differ from one species to another. The different telomere-
405
SECTION 11.5 SUMMARY
Termination of replication in E. coli occurs halfway around the circular chromosome from oriC. Bidirectional replication forks meet head-
on within a terminus region bordered on both sides by multiple Ter sites. Tus binds to Ter and blocks replication forks in one direction but not the other, thus localizing termination to the terminus region. Replication of eukaryotic linear chromosomes cannot be completed at the extreme ends with the replication fork machinery. To solve this end replication problem, telomeres are synthesized at the chromosome ends by telomerase, which carries its own RNA template strand and adds multiple 6-
mer repeats to the 3′ terminus, extending the 3′ single- stranded DNA. The single strand is then converted to duplex DNA by priming and chain extension. After telomere synthesis, removal of the last RNA primer still leaves a small single-
strand gap in the DNA that cannot be filled. Because the telomeric repeats are noncoding and can be replaced by further telomerase action, their loss is of no consequence. Somatic cells lack telomerase, and they die when their telomeres become too short. Telomerase is activated in cancer cells, which become immortal and form tumors. Harnessing the activity of telomerase to kill cancer cells or rejuvenate normal but aging somatic cells is an important subject of medical research.
Telomere DNA–
binding proteins protect the ends of chromosomes from nucleases and recombination.
UNANSWERED QUESTIONS
Although we know the major actors that replicate bacterial chromosomes, the mechanics of advancing a replication fork in the highly condensed DNA of a chromosome in the cell still raises several questions. Regulation of the various steps in replication affects cell division and thus is central to preventing uncontrolled cell growth in diseases such as cancer. Control of replication will undoubtedly be an important subject of future studies.
How do replication forks respond to DNA-
bound proteins? Chromosomal DNA contains many DNA- bound proteins, including repressors, transcription activators, and nucleosomes. We know that the replisome can displace and bypass RNA polymerase in E. coli, but only if the direction of replication is the same as the direction of transcription. What happens when the replication and transcription machineries collide head- on? In eukaryotes, do nucleosomes stay bound to DNA during replication, and how is the epigenetic information contained within them sustained in the daughter chromosomes? What protein modifications control replication? The impact of protein modifications on replication control in eukaryotic cells is extremely important and probably involves the phosphorylation of replication proteins, because their phosphorylation state can be seen to change with phases of the cell cycle. The identity of the kinases, which proteins and amino acid residues they modify, and the change in activity these modifications bring about are nearly unexplored territory.
What is the relationship between telomerase, aging, and cancer? The loss of telomeres leads to chromosome instability and cell death. Most normal somatic cells lack telomerase and die when their telomeres become too short. Immortal cancer cells express telomerase, and their telomeres are maintained. These observations imply that telomerase, aging, and immortality are related. The clinical ramifications of controlling telomere length in cells is a highly active area of current research.
406
HOW WE KNOW: DNA Polymerase Reads the Sequence of the DNA Template to Copy the DNA
Bessman, M.J., I.R. Lehman, E.S. Simms, and A. Kornberg. 1958. Enzymatic synthesis of deoxyribonucleic acid: II. General properties of the reaction. J. Biol. Chem. 233:171–
Lehman, I.R., M.J. Bessman, E.S. Simms, and A. Kornberg. 1958. Enzymatic synthesis of deoxyribonucleic acid: I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J. Biol. Chem. 233:163–

Arthur Kornberg did not intend to discover how DNA was made, or even to become a scientist. He was a physician on a naval ship, but soon after setting out to sea, his single publication as a medical student led to an offer to transfer to the National Institutes of Health. After jumping ship, he began an incredible scientific odyssey that founded the field of replication enzymology.
Kornberg and his group wanted to understand how the DNA polymer was made. They developed an assay for DNA synthesis using bacterial cell extracts to which they added [14C]thymidine to ensure that any radioactive polymer recovered would be DNA and not RNA. Though radioactive incorporation was feeble, it was reproducible. During fractionation of the extract, Kornberg’s group discovered that several heat-
On characterizing Pol I, the researchers were initially puzzled that it required all four dNTPs for robust DNA synthesis. If DNA were serving only as a primer, why couldn’t a DNA polymer be made from just one, two, or three types of nucleotides? The finding implied that the enzyme received instructions from existing DNA acting as a template, as suggested by Watson and Crick, but at that time, the idea of an enzyme receiving direction from its substrate was preposterous. Kornberg’s group conducted experiments to test whether this was in fact the case. They tested DNAs that varied in A=T versus G≡C content, and the result was astounding. Regardless of the mix of dNTPs, the ratio of A=T and G≡C in the product matched that in the template DNA. That settled it! The DNA was serving not only as primer but also as a template. To support this conclusion, they used Pol I to convert the 5.4 kb single-
Then came a discovery by John Cairns that polA mutant E. coli cells, with less than 1% residual Pol I activity, had no growth defects. This result, combined with identification of numerous genes required for replication, revealed a process far more complex than anyone had imagined. Unsettling to Kornberg and his colleagues was the questioning of their work on DNA polymerase I in pointed editorials in Nature New Biology. Did the assays used to purify Pol I result in a red herring? Do “real” polymerases need a primed template? Are dNTPs the true precursors of DNA? Is a 3′→5′ exonuclease proofreader needed by the “real” DNA polymerase?
Kornberg’s son Tom identified both Pol II and Pol III from extracts of polA mutant cells. These polymerases were just like Pol I in the use of a primed template and dNTPs and the presence of a 3′→5′ proofreading exonuclease. Fortunately, the controversial issues raised in Nature New Biology soon vanished. Coincidentally, so did the journal itself.
407
Polymerase Processivity Depends on a Circular Protein That Slides along DNA
Kong X.P., R. Onrust, M. O’Donnell, and J. Kuriyan. 1992. Three-
Stukenberg, P.T., P.S.-V. Studwell, and M. O’Donnell. 1991. Mechanism of the sliding β clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 266:11,328–
DNA polymerases that replicate chromosomes were long known to require “accessory proteins” that somehow confer rapid and processive polymerase activity. However, it seemed a contradiction that proteins that increase the affinity of polymerase for DNA also enable rapid motion along DNA. Specifically, how can a polymerase bind DNA tightly and also rapidly slide along it?
Surprisingly, experiments showed that the β subunit of Pol III holoenzyme, by itself, binds to DNA. This required the γ complex and ATP. However, the β subunit bound only circular DNA and could not bind linear primed DNA. This suggested that the β subunit binds DNA by encircling it, and thus slides off linear DNA. No protein was known to encircle DNA at the time, so this idea was not taken seriously. However, the test was rather simple. A [3H]β subunit was loaded onto circular primed DNA, and the reaction mixture was divided. In one tube, the DNA was linearized using BamHI, and in the other tube, the DNA was untreated and remained circular. The two reaction mixtures were then analyzed on gel filtration columns. The [3H]β bound to the large DNA molecule eluted much earlier (fractions 7 to 16) than [3H]β not bound to DNA (fractions 20 to 40).
If the [3H]β subunit encircles DNA like a doughnut, it should slide off linear DNA but remain on circular DNA. This was exactly the result observed. The solid circles in the upper plot in Figure 1 show the sample treated with BamHI. Most of the [3H]β in this sample eluted late, as [3H]β not associated with DNA. In the untreated sample (open circles), the early fractions show [3H]β bound to DNA. The result is clear: β remains on circular DNA but falls off linear DNA. This behavior suggests that β is shaped like a ring.
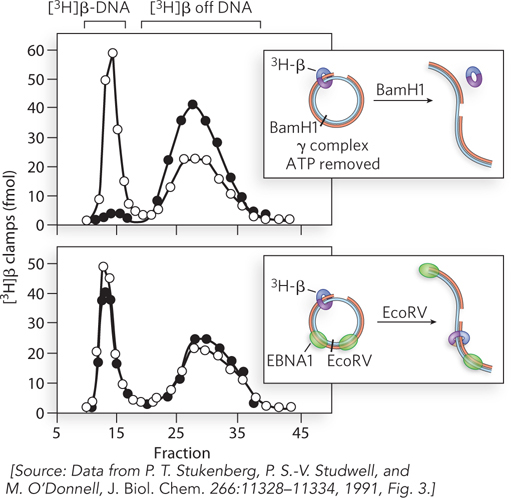
This hypothesis was tested using DNA with two sites for a DNA-
408
HOW WE KNOW: Replication Requires an Origin
Hiraga, S. 1976. Novel F prime factors able to replicate in Escherichia coli Hfr strains. Proc. Natl. Acad. Sci. USA 73:198–
Oka, A., H. Sasaki, K. Sugimoto, and M. Takanami. 1984. Sequence organization of replication origin of the Escherichia coli K-
Zyskind, J.H.W., J.M. Cleary, W.S. Brusilow, N.E. Harding, and D.W. Smith. 1983. Chromosomal replication origin from the marine bacterium Vibrio harveyi functions in Escherichia coli: oriC consensus sequence. Proc. Natl. Acad. Sci. USA 80:1164–
To identify the replication origin of a host cell, plasmid, or virus, a plasmid with a known origin and a selectable marker (e.g., the gene conferring resistance to the antibiotic ampicillin) is first treated with restriction enzymes to excise the origin (Figure 2). DNA extracted from host cells is cut with a restriction enzyme to produce many fragments. Individual fragments are inserted into the cut plasmid, and DNA ligase is used to recreate plasmid DNA circles. These recombinant plasmids are transferred into E. coli, and the transformed cells are plated on selective media (e.g., plates containing ampicillin). To survive the antibiotic in the medium and form a colony, cells must contain the plasmid with the ampicillin-
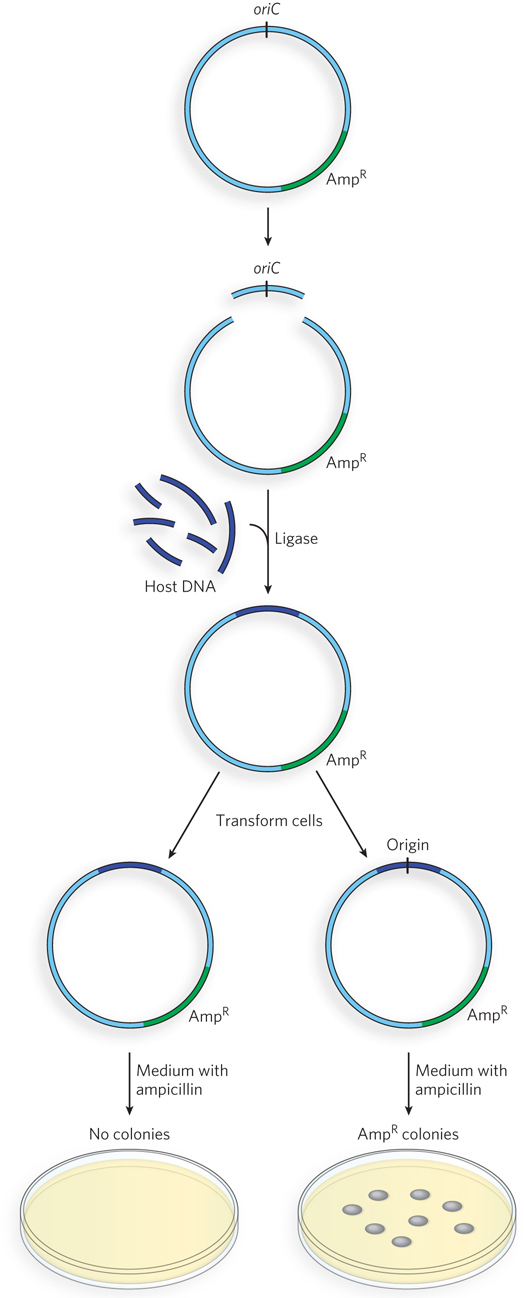
The recombinant plasmid approach has identified numerous origins of bacterial chromosomes, plasmids, and bacteriophage. Yeast (a eukaryote) has defined origins that can be isolated in a similar way. However, eukaryotic plasmids cannot be selected using antibiotics. Instead, genes needed for the metabolism of a particular amino acid are used, and cells are plated on media lacking that amino acid. This experimental approach has not been successful in identifying replication origins in eukaryotes more complex than yeast. It is possible that higher eukaryotes do not have defined origins and that chromatin structure defines replication start sites. Alternatively, the origins of higher eukaryotes are too large to be determined by this method.
409