12.1 TYPES OF DNA MUTATIONS
A mutation is a change in a DNA sequence that is propagated through cellular generations. Mutations can be as small as a single base pair or can range from a few base pairs to thousands. Mutations of one or a few base pairs usually result from errors in replication or damaged nucleotides. Those that span large sections of DNA are typically due to chromosomal rearrangements that arise from errant recombination.
Mutations can have different effects on gene structure and function. A mutation in a gene product can result in a loss of function or a gain of function. For example, loss of function can be the result of mutations that destroy the active site of an enzyme, produce a truncated protein, or disrupt the regulation of gene expression. Gain-of-function mutations might increase the affinity of an enzyme for its substrate, remove the regulatory portion of a protein, or increase gene expression and thus produce more protein. Loss-of-function mutations are generally recessive in a diploid organism, whereas gain-of-function mutations are often dominant over the wild-type allele. Examples of every type of mutation can be observed in human disease. But mutations are also important to life as we know it. Mutations that duplicate entire genes can eventually lead to entirely new proteins with different functions. Mutations have given us new, higher yields of grain. And mutations also lead to new species, providing the diversity that drives evolution. Without mutations you would not be here.
A Point Mutation Can Alter One Amino Acid
A change in a single base pair is often referred to as a point mutation. Point mutations fall into two categories depending on the types of base substitution in the DNA sequence (Figure 12-1). A transition mutation is the exchange of a purine-pyrimidine base pair for the other purine-pyrimidine base pair: C≡G becomes T=A, or T=A becomes C≡G. A transversion mutation is the replacement of a purine-pyrimidine base pair with a pyrimidine-purine base pair, or vice versa. For example, C≡G becomes either G≡C or A=T. Transition mutations exchange bases that have a similar size, which is easier for a polymerase to accommodate, and thus transitions are nearly 10 times more frequent than transversions. A point mutation in the protein-coding region of a gene can result in an altered protein with partial or complete loss of function. If the protein is central to cell viability, the cell could die.
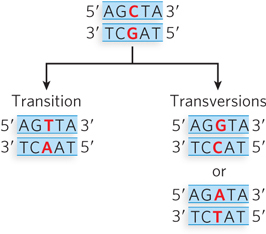
Figure 12-1: Transition and transversion point mutations. The parental DNA (top) contains a C≡G base pair. There are two possible point mutations: a transition (left), in which a purine (in this case, G) is replaced with a different purine (A), producing a T=A base pair on replication; or a transversion (right), in which a pyrimidine (in this case, C) is replaced with a purine (G or A) to produce either a G≡C or an A=T base pair. (To review hydrogen bonding between base pairs, see Figure 1-3.)
Point mutations in a protein-coding region can be classified by their effect on the protein sequence. The DNA sequence encoding a protein is read in triplets, or codons. Each codon corresponds to an amino acid (see Chapter 17). A silent mutation is a nucleotide change that produces a codon for the same amino acid. For example, GAA and GAG both code for glutamate. A missense mutation is a nucleotide change that results in a different amino acid, such as a change from glutamate (GAA) to glutamine (CAA). A nonsense mutation changes the nucleotide sequence so that instead of encoding an amino acid, the triplet functions as a stop codon, terminating the translation process and generating a truncated protein without a complete amino acid sequence.
KEY CONVENTION
When a point mutation results in an altered protein sequence, the change in that protein’s amino acid sequence is denoted by letters and a number—for example, E214A. The first letter is the single-letter abbreviation for the amino acid residue in the wild-type protein (E); the number is the position of the residue, numbering from the N-terminus of the protein sequence (214); and the second letter is the amino acid residue in the mutant protein (A). A nonsense mutation is identified by an X as the second letter—for example, E214X.
Point mutations are produced in a two-step process. In the first step, an incorrect nucleotide is incorporated by a DNA polymerase (Figure 12-2). This can happen when DNA polymerase encounters a damaged base in the template strand that no longer forms a normal base pair. It can also occur without DNA damage, because DNA polymerases, despite their high accuracy, sometimes make a mistake. For example, as discussed in Chapter 11, a DNA polymerase can incorporate a tautomeric form of a dNTP, resulting in a mismatch. Regardless of how the mismatch is formed, it is not yet a mutation, because it can be detected and repaired by processes described later in this chapter. However, if the mismatch is not repaired, it becomes a mutation in a second step during replication, which incorporates the mismatch into a fully base-paired duplex DNA. This results in a new, correctly paired base pair that can no longer be detected by repair enzymes but alters the original sequence and is therefore an inheritable mutation.
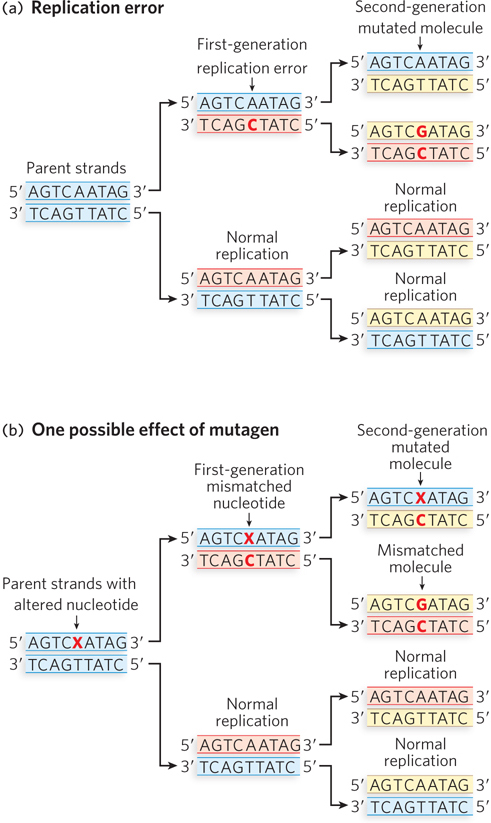
Figure 12-2: The two-step process of a point mutation. (a) DNA polymerase incorporates an incorrect nucleotide (C instead of T), leading to a mismatch in one of the first-generation cells. If the mismatch is not corrected before this cell replicates, one of the second-generation cells will incur a mutation. (b) The parental DNA contains a damaged adenosine base (X). The damaged A more readily pairs with a C than a T, so replication over the damaged base results in a mismatched nucleotide in one of the first-generation cells. Further replication can result in a permanent mutation in a cell of the second generation.
Evolution depends on mutations that can confer a selective advantage, but it is relatively rare for a mutation to have a positive outcome for the organism. Studies in Drosophila melanogaster suggest that mutations that alter the protein sequence are most likely to be harmful; about 70% have a negative effect, and the rest either are neutral or have a weak beneficial effect. As shown by studies in yeast, however, for mutations outside the protein-coding region, fewer than 7% are harmful.
Point mutations are known to cause a wide variety of human diseases. One example is sickle-cell anemia (discussed in Highlight 2-1), resulting from a transversion that produces an amino acid change in hemoglobin: a GAG, encoding glutamate (E), at residue 6 changes to GTG, encoding valine (V) (E6V). Another example of a point mutation in human disease is Werner syndrome, which causes premature aging due to genetic instability. The WRN gene encodes a helicase. Several different point mutations in the WRN gene lead to Werner syndrome. Some are of the missense type (e.g., K32R), although most are nonsense mutations that lead to a shorter protein product (e.g., E41X).
The most harmful mutations are those occurring in the genes involved in DNA repair, because these often result in cancer. As we discuss later in this chapter, the repair of mismatch errors requires many different proteins. A mutation in a gene encoding a mismatch repair protein can result in the production of many more mutations in the cell, as subsequent mutations are no longer corrected. The occurrence of many mutations in a cell can result in cancer, because, eventually, a mutation will occur in a gene (or genes) that encodes a protein needed to control cell division. In normal cells, oncogenes encode proteins that drive the cell division cycle forward, and tumor suppressor genes encode proteins that suppress cell division. Many tumor suppressors are transcription factors that regulate the expression of genes that drive the cell cycle. The transcription factor p53 and the retinoblastoma protein are examples of tumor suppressors that are mutated in many types of cancer.
Small Insertion and Deletion Mutations Change Protein Length
Another type of mutation is the gain or loss of one or more base pairs. Insertion mutations occur when one or more base pairs are added to the wild-type sequence; conversely, deletion mutations are due to the loss of one or more base pairs. Insertion and deletion mutations are collectively referred to as indels. Indels are caused by aberrant recombination or by template slippage by the DNA polymerase during replication. As we discuss in more detail in Chapter 17, proteins are encoded (via a messenger RNA intermediary) in a series of codons that starts with ATG and ends with one of three different codons that signal translation to stop (Figure 12-3a). The protein is synthesized from its N-terminus to its C-terminus. The DNA sequence from the start codon to the stop codon is referred to as a reading frame. Because nucleotides are decoded in triplets, an indel mutation of only one or two base pairs in the coding sequence of a protein throws off the reading frame after the mutation, resulting in a frameshift mutation (Figure 12-3b). Given the frequency of stop codons in a random sequence (1 in every 20 codons), these mutations usually produce a truncated protein, especially if the gene is large and the mutation occurs a few hundred base pairs prior to the wild-type stop codon. Frameshift mutations often destroy the protein’s function, especially if the truncation is close to the N-terminus.

Figure 12-3: Insertions and deletions can lead to frameshift mutations. (a) A reading frame in the DNA begins with the start codon, ATG, and stops with a stop codon (TAG in this case). When the coding strand of the DNA is transcribed into mRNA, these sequences are AUG (start) and UAG (stop). (b) Insertions or deletions of one or two base pairs in a protein-coding sequence result in frameshift mutations, in which the reading frame becomes out of register relative to the wild-type sequence and typically leads to a premature stop codon.
Indel mutations that occur in multiples of three base pairs preserve the reading frame of the gene. The most common such mutation is an insertion of three nucleotides. This is thought to be caused by template slippage during replication, with the inserted triplet embedded in a region of triplet repeats (Figure 12-4). Thus, the wild-type gene has a repeating array of codons, and the protein product contains a region of repeats of the same amino acid. Although the mutant protein, with an added amino acid residue, is slightly larger than the wild-type protein, it usually retains some degree of function.
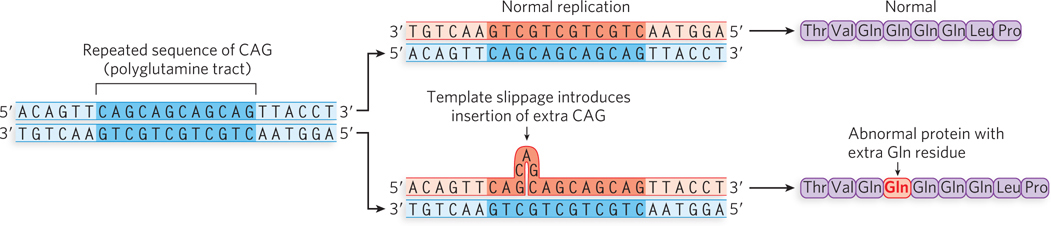
Figure 12-4: A three-nucleotide insertion mutation. Template slippage on repeated sequences during replication can lead to small insertions and deletions. Shown here is a three-nucleotide (CAG) insertion in a repeated CAG region of the coding strand of a gene sequence, resulting in an additional Gln residue in a polyglutamine tract. The protein sequence shown is the sequence encoded by the coding strand of DNA, the strand that is transcribed into mRNA.
There are many examples of human disease caused by the insertion of triplet sequences, often referred to as triplet expansion diseases (Table 12-1). More than half of the triplet expansion diseases involve expansion of the CAG codon for glutamine (Q) and are known as polyglutamine (polyQ) diseases. An example is Huntington disease. When the number of CAG repeats increases to 36 copies or more, degeneration of the cerebral cortex may occur in midlife (see Figure 2-28). The number of triplet repeats correlates with the severity and time of onset of this neurological disorder.
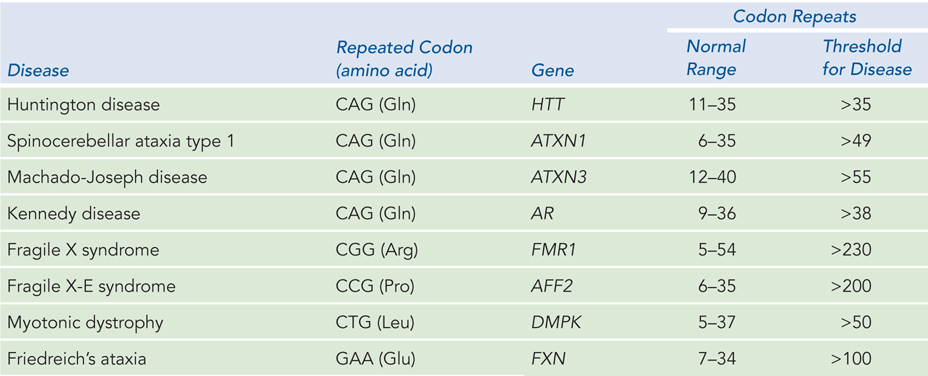
Figure 12-1: Triplet Expansion Diseases
The first triplet expansion disease to be identified was fragile X syndrome, which causes a form of mental impairment. The defect in fragile X syndrome maps to the X chromosome and involves a CGG repeat. Humans normally have 5 to 54 repeats, but some individuals have more than 230 repeats (up to 2,000), and this leads to the fragile X mental impairment. Individuals with 54 to 230 repeats show no symptoms but are carriers of the disease, because the genetic instability leading to the triplet expansion occurs in germ-line cells and therefore becomes worse with each generation—until the threshold of 230 is passed and the disease manifests. Diagnostic genetic screening for triplet expansion diseases can be performed with PCR primers that anneal to the unique sequences known to lie on either side of the repeated region (Figure 12-5).
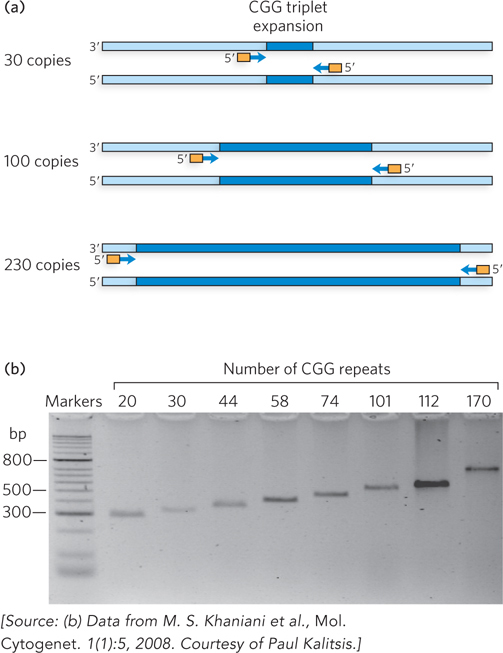
Figure 12-5: PCR-based genetic testing for fragile X syndrome, a triplet expansion disease. (a) The number of repeats in the fragile X locus can be determined by length analysis of PCR products. This uses PCR primers that hybridize to unique sequences bordering the region of interest, then the PCR products are analyzed by gel electrophoresis. The size of a PCR product correlates with the size of the repeated region in the gene. (b) Size of the repeated unit in the gene for eight different patients.
Some Mutations Are Very Large and Form Abnormal Chromosomes
Some types of mutations involve extensive changes in the DNA sequence, most commonly caused by aberrant recombination events (Figure 12-6a). These types of mutations generally cannot be repaired, but are mentioned here because they form an important class of mutations that cause disease, and they also drive the process of evolution. One type of mutation that changes an extensive amount of DNA is the deletion of a large tract of DNA that can lead to a complete loss of genes and also bring genes into proximity that were once far apart. The opposite of a deletion is a duplication mutation, the amplification of a large tract of DNA, leading to increased gene dosage effects (i.e., increased amounts of product from the amplified gene). Chromosome inversion mutations result from the inversion of a large section of DNA in a chromosome and can have varied effects, especially on the genes at the break points. Aberrant recombination events can also occur between two different chromosomes. For example, a region of DNA from one chromosome can be transferred as an insertion to another chromosome. A chromosome translocation mutation occurs when two nonhomologous chromosomes exchange large regions of DNA. In particularly rare instances, entire chromosomes may be fused to each other. Such large rearrangements are almost always deleterious and almost never inherited. However, when the product is viable and is inherited, it can have major evolutionary consequences. A fusion of two simian chromosomes was an important event in the evolution of hominids (Figure 12-6b).
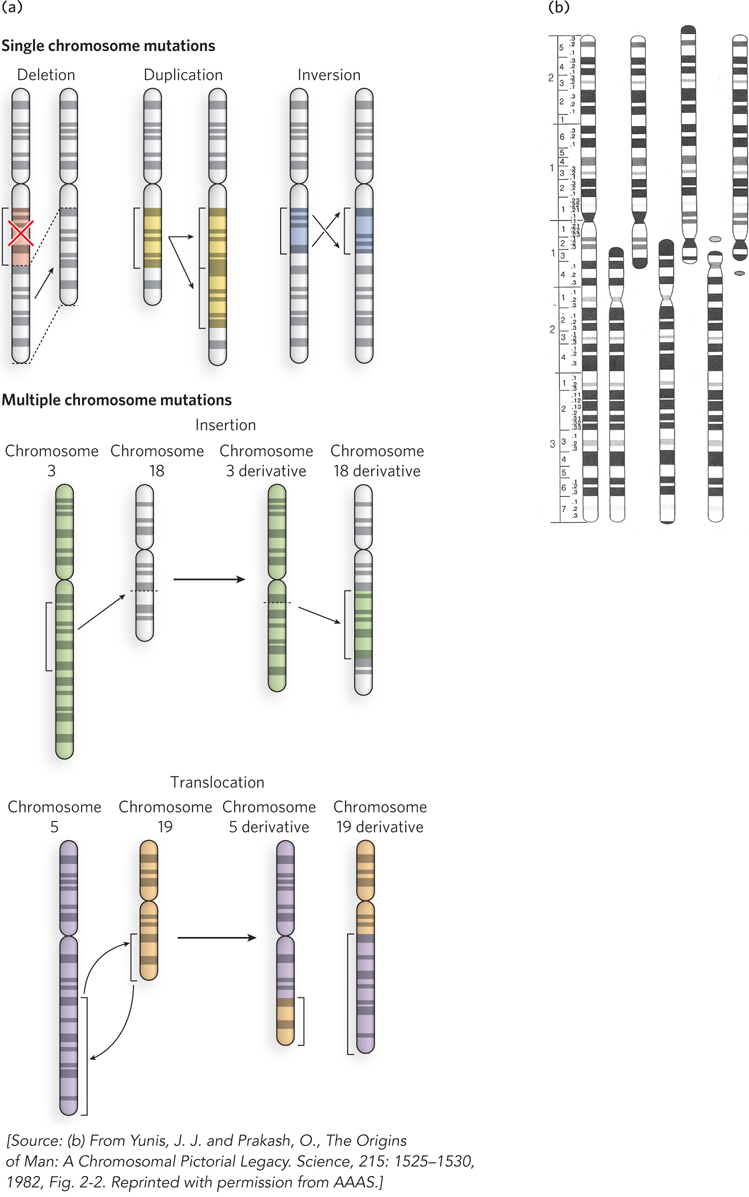
Figure 12-6: Large-scale mutations. (a) Mutations that lead to alterations in a chromosome can occur internally through deletion, duplication, or inversion events (top), or they can be due to an insertion (middle) or translocation (bottom), exchanging DNA with another chromosome. (b) Simians such as chimpanzees have a genome consisting of 25 different chromosomes, rather than the 24 different chromosomes (22 autosomes plus the sex chromosomes X and Y) found in humans. The distinction is not as great as it sounds. Two simian chromosomes were fused during the evolution of hominids, producing human chromosome 2—shown next to the related chromosomes from (left to right) chimpanzee, gorilla, and orangutan.
Chromosomal abnormalities can result in the formation of a fusion gene, a hybrid of two different genes. Several types of fusion genes are associated with various forms of cancer, including lymphoma, sarcoma, and prostate cancer. An example of a chromosomal translocation that forms a carcinogenic fusion gene is the formation of BCR-ABL, in which one end of chromosome 9 breaks and rejoins (i.e., recombines) with part of chromosome 22, forming a very small derivative chromosome 22 (Figure 12-7). The break point in chromosome 9 occurs within the ABL gene, which becomes fused with the BCR gene in chromosome 22, forming the BCR-ABL fusion gene. The ABL protein is a cell cycle protein kinase, a tyrosine kinase that helps control the cell cycle by phosphorylating certain proteins (on specific Tyr residues) in response to cellular signals. When ABL becomes fused with BCR, the sections of the ABL protein that regulate the kinase activity are lost; the resulting unregulated tyrosine kinase activity leads to the uncontrolled cell division associated with lymphoblastic leukemia.
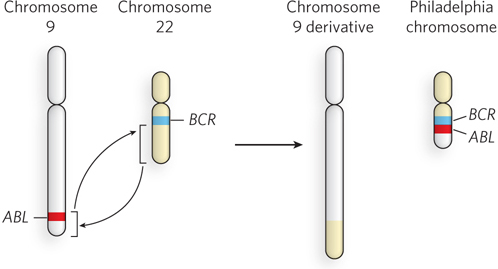
Figure 12-7: A chromosomal translocation resulting in a fusion gene. The chromosomal translocation that fuses the ABL gene with the BCR gene occurs when a piece of chromosome 9 (with ABL) is exchanged with a piece of chromosome 22. The BCR-ABL fusion gene causes leukemia. The small chromosome resulting from the translocation (the chromosome 22 derivative), which carries the fusion gene, is referred to as the Philadelphia chromosome, after the city where the translocation was first identified and studied.
Sometimes a fusion gene retains production of the normal protein but comes under the control of a strong, unregulated promoter that produces too much of the protein. An example involves the C-MYC gene, an oncogene, on chromosome 8. A common translocation involving this gene, found in certain types of cancer, occurs between chromosomes 8 and 14.
SECTION 12.1 SUMMARY
A mutation is a change in DNA sequence. It can be a point mutation, affecting a single base pair, an insertion or deletion affecting more than one base pair, or a chromosomal rearrangement that affects many genes on the chromosome.
Point mutations are classified as transitions and transversions. A transition converts a purine-pyrimidine base pair to the other purine-pyrimidine pair, or pyrimidine-purine to pyrimidine-purine. A transversion converts a purine-pyrimidine base pair to a pyrimidine-purine pair, or vice versa.
Most mutations are produced in a two-step process. First, a nucleotide is either damaged or misincorporated during replication, then subsequent replication pairs an incorrect nucleotide with the damaged or misincorporated nucleotide.
Most mutations are deleterious; for example, mutations in oncogenes and tumor suppressor genes that control the cell division cycle can lead to cancer.
Insertion and deletion mutations are the addition and removal of nucleotides in a DNA sequence. Insertions and deletions that are not multiples of three nucleotides can shift a gene’s reading frame, resulting in at runcated protein. Triplet expansion diseases result from three-base-pair insertion mutations caused by template slippage during replication.
Large-scale mutations can produce abnormal chromosomes; they occur when parts of chromosomes are deleted, duplicated, inverted, or exchanged. These mutations can form fused genes, some of which cause cancer.