14.3 Bioinformatics: Meaning from Genomic Sequence
The genomic sequence is a highly encrypted code containing the raw information for building and operation of organisms. The study of the information content of genomes is called bioinformatics. We are far from being able to read this information from beginning to end in the way that we would read a book. Even though we know which triplets encode which amino acids in the protein-
The nature of the information content of DNA
DNA contains information, but in what way is it encoded? Conventionally, the information is thought of as the sum of all the gene products, both proteins and RNAs. However, the information content of the genome is more complex than that. The genome also contains binding sites for different proteins and RNAs. Many proteins bind to sites located in the DNA itself, whereas other proteins and RNAs bind to sites located in mRNA (Figure 14-9). The sequence and relative positions of those sites permit genes to be transcribed, spliced, and translated properly, at the appropriate time in the appropriate tissue. For example, regulatory protein-
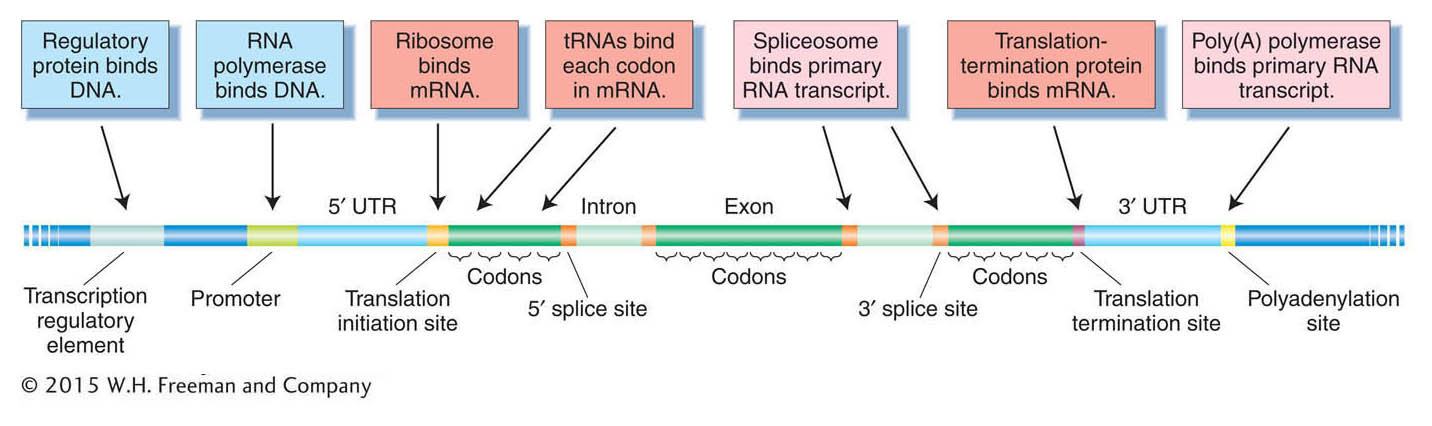
Deducing the protein-encoding genes from genomic sequence
Because the proteins present in a cell largely determine its morphology and physiological properties, one of the first orders of business in genome analysis and annotation is to try to determine an inventory of all of the polypeptides encoded by an organism’s genome. This inventory is termed the organism’s proteome It can be considered a “parts list” for the cell. To determine the list of polypeptides, the sequence of each mRNA encoded by the genome must be deduced. Because of intron splicing, this task is particularly challenging in multicellular eukaryotes, where introns are the norm. In humans, for example, an average gene has about 10 exons. Furthermore, many genes encode alternative exons; that is, some exons are included in some versions of a processed mRNA but are not included in others (see Chapter 8). The alternatively processed mRNAs can encode polypeptides having much, but not all, of their amino acid sequences in common. Even though we have a great many examples of completely sequenced genes and mRNAs, we cannot yet identify 5′ and 3′ splice sites merely from DNA sequence with a high degree of accuracy. Therefore, we cannot be certain which sequences are introns. Predictions of alternatively used exons are even more error prone. For such reasons, deducing the total polypeptide parts list in higher eukaryotes is a large problem. Some approaches follow.
ORF detection The main approach to producing a polypeptide list is to use the computational analysis of the genome sequence to predict mRNA and polypeptide sequences, an important part of bioinformatics. The procedure is to look for sequences that have the characteristics of genes. These sequences would be genesize and composed of sense codons after possible introns had been removed. The appropriate 5′- and 3′-end sequences would be present, such as start and stop codons. Sequences with these characteristics typical of genes are called open reading frames (ORFs). To find candidate ORFs, computer programs scan the DNA sequence on both strands in each reading frame. Because there are three possible reading frames on each strand, there are six possible reading frames in all.
Direct evidence from cDNA sequences Another means of identifying ORFs and exons is through the analysis of mRNA expression. This analysis can be done in two ways. Both methods involve the synthesis of libraries of DNA molecules that are complementary to mRNA sequences, called cDNA (see Chapter 10). The longest established method entails the cloning and amplification of these cDNA molecules in a vector. However, NGS technologies allow for the direct sequencing of short cDNA molecules without the cloning step (called RNA sequencing or “RNA-
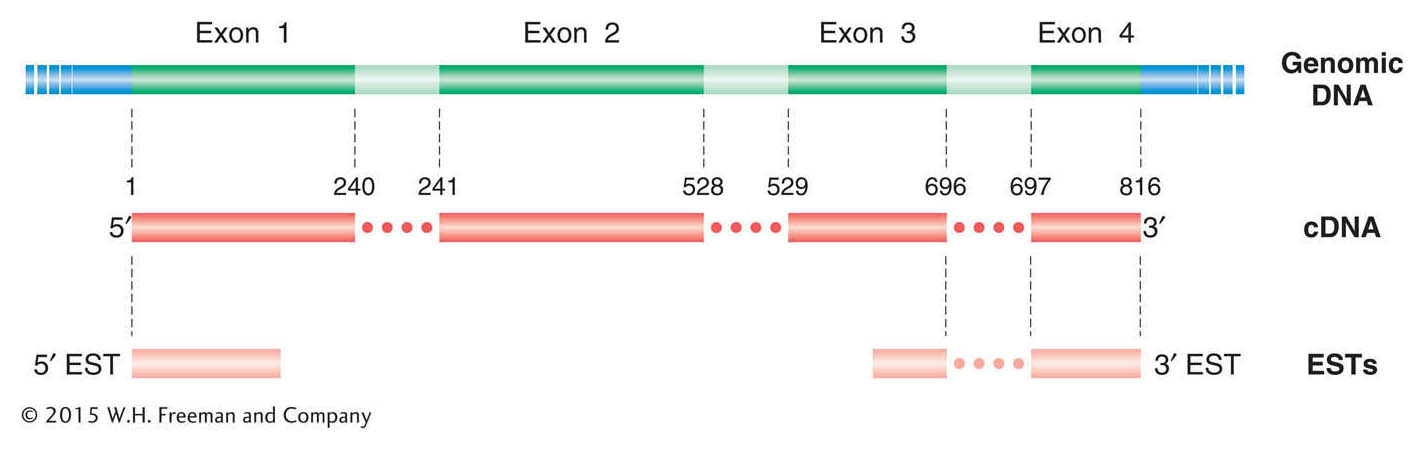
The alignment of cDNAs with their corresponding genomic sequence clearly delineates the exons, and hence introns are revealed as the regions falling between the exons. In the assembled cDNA sequence, the ORF should be continuous from initiation codon through stop codon. Thus, cDNA sequences can greatly assist in identifying the correct reading frame, including the initiation and stop codons. Full-
In addition to full-
Predictions of binding sites As already discussed, a gene consists of a segment of DNA that encodes a transcript as well as the regulatory signals that determine when, where, and how much of that transcript is made. In turn, that transcript has the signals necessary to determine its splicing into mRNA and the translation of that mRNA into a polypeptide (Figure 14-11). There are now statistical “gene-
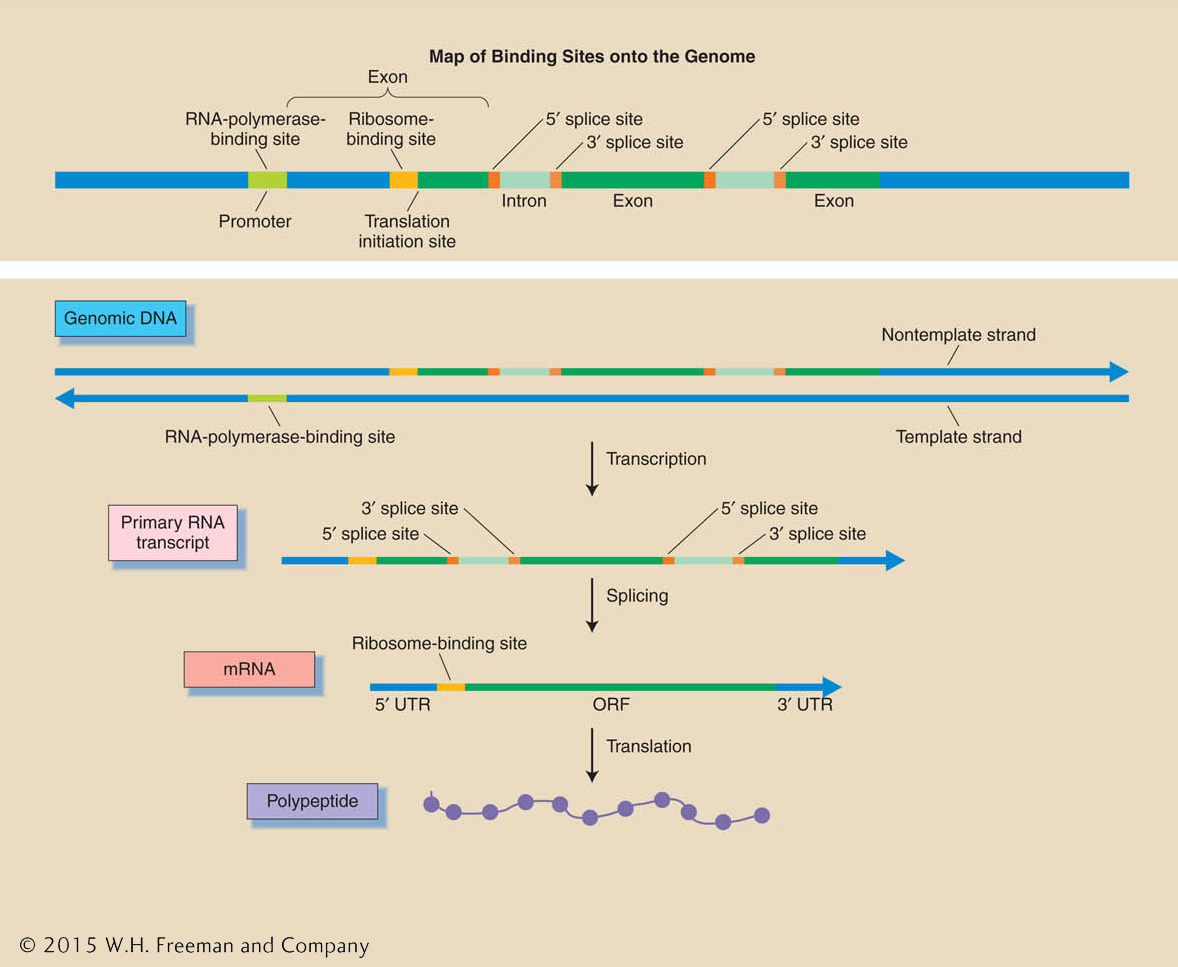
Using polypeptide and DNA similarity Because organisms have common ancestors, they also have many genes with similar sequences in common. Hence, a gene will likely have relatives among the genes isolated and sequenced in other organisms, especially in the closely related ones. Candidate genes predicted by the preceding techniques can often be verified by comparing them with all the other gene sequences that have ever been found. A candidate sequence is submitted as a “query sequence” to public databases containing a record of all known gene sequences. This procedure is called a BLAST search (BLAST stands for Basic Local Alignment Search Tool). The sequence can be submitted as a nucleotide sequence (a BLASTn search) or as a translated amino acid sequence (BLASTp). The computer scans the database and returns a list of full or partial “hits,” starting with the closest matches. If the candidate sequence closely resembles that of a gene previously identified from another organism, then this resemblance provides a strong indication that the candidate gene is a real gene. Less-
BLAST searches are used in many other ways, but always the goal is to find out more about some identified sequence of interest.
Predictions based on codon bias Recall from Chapter 9 that the triplet code for amino acids is degenerate; that is, most amino acids are encoded by two or more codons (see Figure 9-5). The multiple codons for a single amino acid are termed synonymous codons. In a given species, not all synonymous codons for an amino acid are used with equal frequency. Rather, certain codons are present much more frequently in mRNAs (and hence in the DNA that encodes them). For example, in D. melanogaster, of the two codons for cysteine, UGC is used 73 percent of the time, whereas UGU is used 27 percent. This usage is a diagnostic for Drosophila because, in other organisms, this “codon bias” pattern is quite different. Codon biases are thought to be due to the relative abundance of the tRNAs complementary to these various codons in a given species. If the codon usage of a predicted ORF matches that species’ known pattern of codon usage, then this match is supporting evidence that the proposed ORF is genuine.
Putting it all together A summary of how different sources of information are combined to create the best-
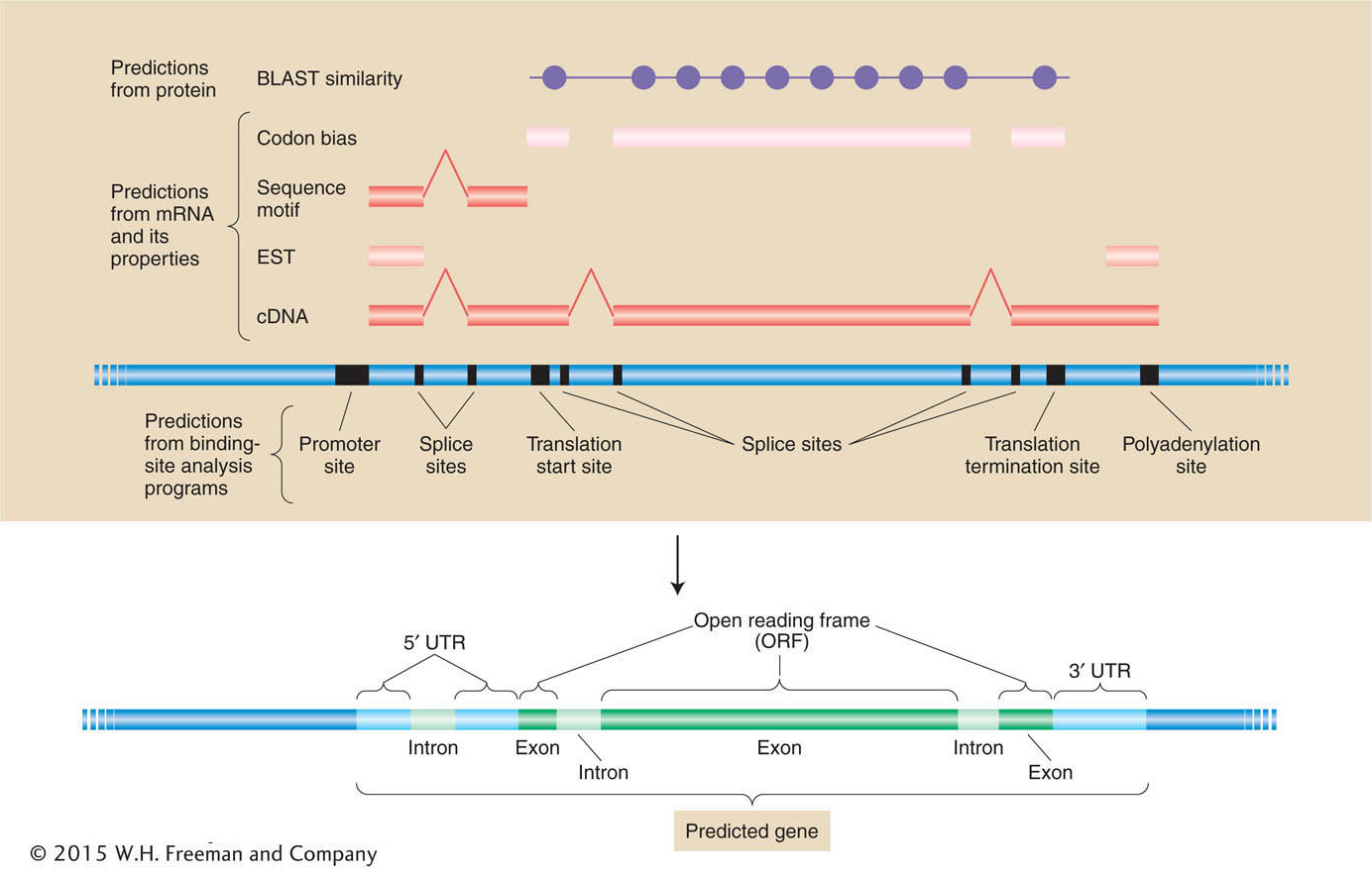
KEY CONCEPT
Predictions of mRNA and polypeptide structure from genomic DNA sequence depend on the integration of information from cDNA sequence, binding-Let’s consider some of the insights from our first view of the overall genome structures and global parts lists of a few species whose genomes have been sequenced. We will start with ourselves. What can we learn by looking at the human genome by itself? Then we will see what we can learn by comparing our genome with others.