14.4 The Structure of the Human Genome
In describing the overall structure of the human genome, we must first confront its repeat structure. A considerable fraction of the human genome, about 45 percent, is repetitive. Much of this repetitive DNA is composed of copies of transposable elements. Indeed, even within the remaining single-copy DNA, a fraction has sequences suggesting that they might be descended from ancient transposable elements that are now immobile and have accumulated random mutations, causing them to diverge in sequence from the ancestral transposable elements. Thus, much of the human genome appears to be composed of genetic “hitchhikers.”
Only a small part of the human genome encodes polypeptides; that is, somewhat less than 3 percent of it encodes exons of mRNAs. Exons are typically small (about 150 bases), whereas introns are large, many extending more than 1000 bases and some extending more than 100,000 bases. Transcripts are composed of an average of 10 exons, although many have substantially more. Finally, introns may be spliced out of the same gene in locations that vary. This variation in the location of splice sites generates considerable added diversity in mRNA and polypeptide sequence. On the basis of current cDNA and EST data, at least 60 percent of human protein-coding genes are likely to have two or more splice variants. On average, there are several splice variants per gene. Hence, the number of distinct proteins encoded by the human genome is several-fold greater than the number of recognized genes.
The number of genes in the human genome has not been easy to pin down. In the initial draft of the human genome, there were an estimated 30,000 to 40,000 protein-coding genes. However, the complex architecture of these genes and the genome can make annotation difficult. Some sequences scored as genes may actually be exons of larger genes. In addition, there are more than 19,000 pseudogenes, which are ORFs or partial ORFs that may at first appear to be genes but are either nonfunctional or inactive due to the manner of their origin or to mutations. So-called processed pseudogenes are DNA sequences that have been reverse-transcribed from RNA and randomly inserted into the genome. Ninety percent or so of human pseudogenes appear to be of this type. About 900 pseudogenes appear to be conventional genes that have acquired one or more ORF-disrupting mutations in the course of evolution. As the challenges in annotation have been overcome, the estimated number of genes in the human genome has dropped steadily. A recent estimate is that there are about 21,000 protein-coding genes.
The annotation of the human genome progressed as the sequences of each chromosome were finished one by one. These sequences then became the searching ground in the hunt for candidate genes. An example of gene predictions for a chromosome from the human genome is shown in Figure 14-13. Such predictions are being revised continually as new data become available. The current state of the predictions can be viewed at many Web sites, most notably at the public DNA databases in the United States and Europe (see Appendix B). These predictions are the current best inferences of the protein-coding genes present in the sequenced species and, as such, are works in progress.
Figure 14-13: The sequence map of human chromosome 20
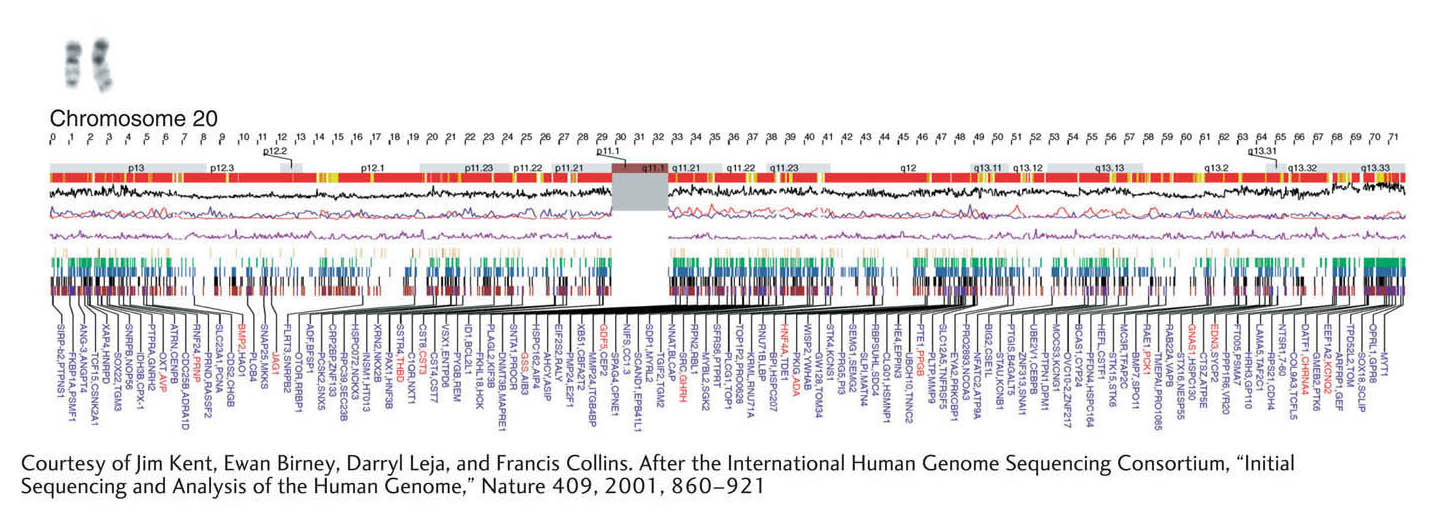
Figure 14-13: Numerous genes have been identified on human chromosome 20. The recombinational and cytogenetic map coordinates are shown in the top lines of the figure. Various graphics depicting gene density and different DNA properties are shown in the middle sections. The identifiers of the predicted genes are shown at the bottom of the panel.
[Courtesy of Jim Kent, Ewan Birney, Darryl Leja, and Francis Collins. After the International Human Genome Sequencing Consortium, “Initial Sequencing and Analysis of the Human Genome,” Nature 409, 2001, 860–921.]
Page 525
Noncoding functional elements in the genome
The discussion thus far has focused exclusively on the protein-coding regions of the genome. This emphasis is due more to analytical ease than to biological importance. Because of the simplicity and universality of the genetic code, and the ability to synthesize cDNA from mRNA, the detection of ORFs and exons is much easier than the detection of functional noncoding sequences. As stated earlier, only 3 percent of the human genome encodes exons of mRNAs, and fewer than half of these exon sequences, a little over 1 percent of the total genome DNA, encode protein sequences. So, more than 98 percent of our genome does not encode proteins. How do we identify other functional parts of the genome?
Introns and 5′ and 3′ untranslated sequences are readily annotated by analysis of gene transcripts, while gene promoters are usually identified by their proximity to transcription units and signature DNA sequences. However, other regulatory sequences such as enhancers are not identifiable by mere inspection of DNA sequences, and other sequences that encode various kinds of RNA transcripts (microRNAs, small interfering RNAs, long noncoding RNAs) require detection and annotation of their transcripts. While many such noncoding elements have been identified in the course of the study of human molecular genetics, the potentially vast number of such elements warrants a more systematic approach. The Encyclopedia of DNA Elements (ENCODE) project was thus launched with the ambitious goal of identifying all functional elements within the human genome.
Page 526
This large-scale collaborative endeavor has employed a diverse array of techniques to detect sequences potentially involved in the control of gene transcription, as well as all transcribed regions. Because such sequences are expected to be active in only individual or subsets of cell types, researchers studied 147 human cell types. By searching for regions that were associated with the binding of transcription factors, the ENCODE project estimated that there are approximately 500,000 potential enhancers associated with known genes. The project also detected transcripts emanating from 80 percent of the human genome.
This is a much larger fraction of the genome than was expected. After all, as stated above, only a little over 1 percent of the genome is coding sequence. However, the production of a transcript does not necessarily mean that the transcript contributes to human biology. It is possible that some proportion of these transcripts represent “noise” in the cell—transcripts that have no biological function, but also do no harm. It is not sound to ascribe function to a sequence without some form of additional data, so what kinds of additional data can be used to resolve questions of function?
Evolutionary conservation of sequences has proven to be a good indicator of biological function. Sequences will not be preserved over evolutionary time unless mutations that alter them are weeded out by natural selection. One way to locate potentially functional noncoding elements then is to look for conserved sequences, which have not changed much over millions of years of evolution.
For example, one can search for very highly conserved sequences of modest length among a few species or for less perfectly conserved sequences of greater length among a larger number of species. Comparisons of the human, rat, and mouse genomes have led to the identification of so-called ultraconserved elements, which are sequences that are perfectly conserved among the three species. Searches of these genomes have found more than 5000 sequences of more than 100 bp and 481 sequences of more than 200 bp that are absolutely conserved. Although many of these elements are found in gene-poor regions, they are most richly concentrated near regulatory genes important for development. The majority of highly conserved noncoding elements may largely take part in regulating the expression of the genetic toolkit for the development of mammals and other vertebrates (see Chapter 13).
How can we verify that such conserved elements play a role in gene regulation? These elements can be tested in the same manner as the transcriptional cis-acting regulatory elements examined in earlier chapters, with the use of reporter genes (see Section 12.2). A researcher places candidate regulatory regions adjacent to a promoter and reporter gene and introduces the reporter gene into a host species. One such example is shown in Figure 14-14. An element that is highly conserved among mammalian, chicken, and a frog species lies 488 kb from the 3′ end of the human ISL1 gene, which encodes a protein required for motor-neuron differentiation. This element was placed upstream of a promoter and the ß-galactosidase (lacZ) reporter gene, and the construct was injected into the pronuclei of fertilized mouse oocytes. The reporter protein is then seen to be expressed along the spinal cord and in the head, as one would expect for the location of future motor neurons (see Figure 14-14). Most significantly, the expression pattern corresponds to part of the expression pattern of the native mouse ISL1 gene (presumably other noncoding elements control the other features of ISL1 expression). The expression pattern strongly suggests that the conserved element is a regulatory region for the ISL1 gene in each species. Many thousands of human noncoding regulatory elements will likely be identified on the basis of sequence conservation and the activity of those elements in reporter assays.
KEY CONCEPT
The noncoding functional elements of the genome are much more difficult to identify than coding sequences, and require a combination of comparative and experimental evidence to validate.
Figure 14-14: Testing the role of a conserved element in gene regulation
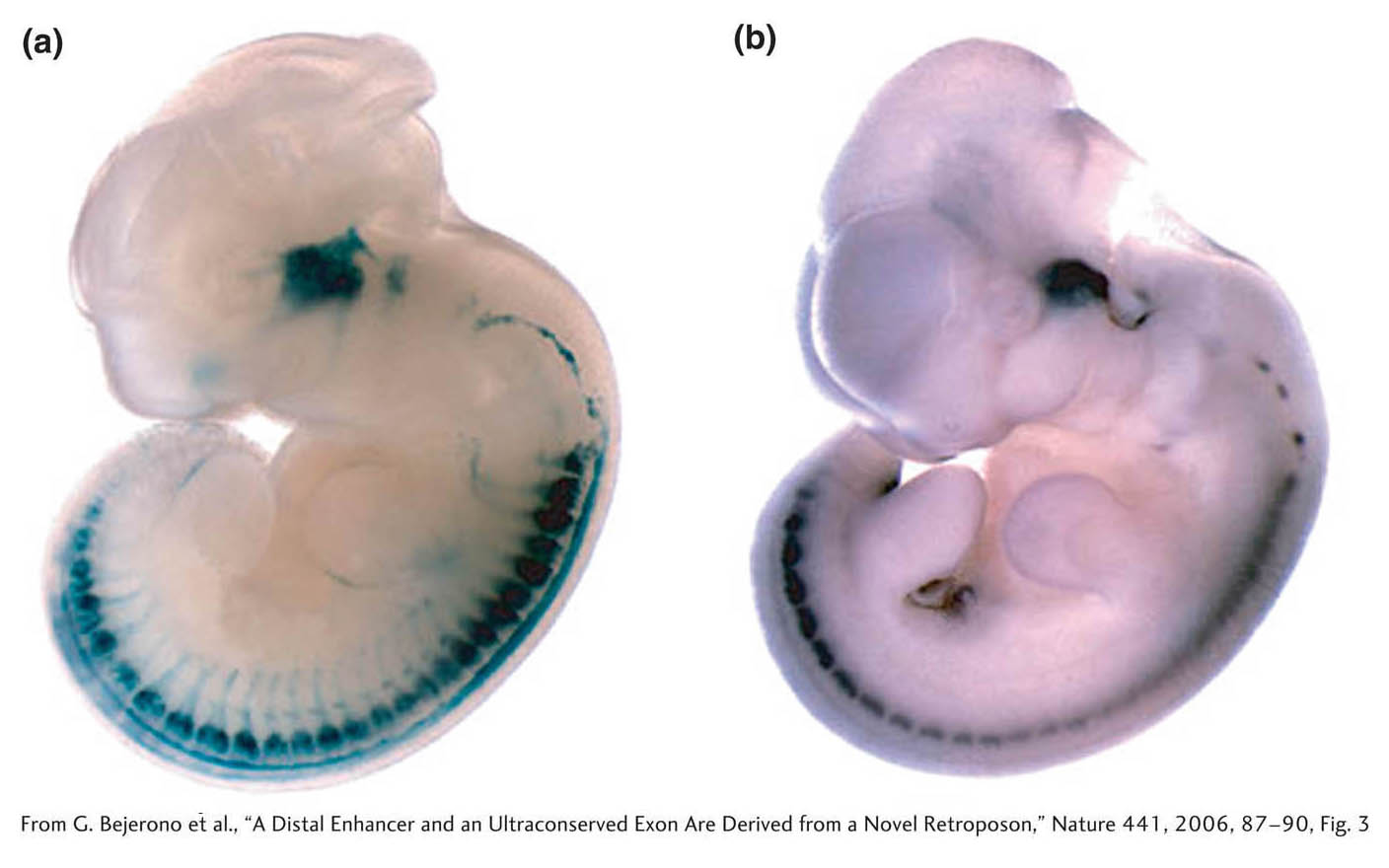
Figure 14-14: A transcriptional cis-acting regulatory element is identified in an ultraconserved element of the human genome. An ultraconserved element lying near the human ISL1 gene was coupled to a reporter gene and injected into fertilized mouse oocytes. The regions where the gene is expressed are stained dark blue or black. (a) The reporter gene is expressed in the head and spinal cord of a transgenic mouse, as seen here on day 11.5 of gestation. This expression pattern corresponds to (b) the native pattern of expression of the mouse ISL1 gene on day 11.5 of gestation. This experiment demonstrates how functional noncoding elements can be identified by comparative genomics and tested in a model organism.
[From G. Bejerono et al., “A Distal Enhancer and an Ultraconserved Exon Are Derived from a Novel Retroposon,” Nature 441, 2006, 87–90, Fig. 3.]
Page 527