The ABC Cystic Fibrosis Transmembrane Regulator Is a Chloride Channel, Not a Pump
Several human genetic diseases are associated with defective ABC proteins (see Table 11-3). The best-studied and most widespread is cystic fibrosis (CF), caused by a mutation in the gene encoding the cystic fibrosis transmembrane regulator (CFTR, also called ABCC7). Like other ABC proteins, CFTR has two transmembrane T domains and two cytosolic A, or ATP-binding, domains. CFTR contains an additional R (regulatory) domain on the cytosolic face; R links the two homologous halves of the protein, creating an overall domain organization of T1–A1–R–T2–A2. But CFTR is a Cl− channel, not a pump. It is expressed in the apical plasma membranes of epithelial cells in the lungs, sweat glands, pancreas, and other tissues. For instance, CFTR protein is important for reuptake into the cells of sweat glands of Cl− lost by sweating; babies with cystic fibrosis, if licked, often taste “salty” because this reuptake is inhibited.
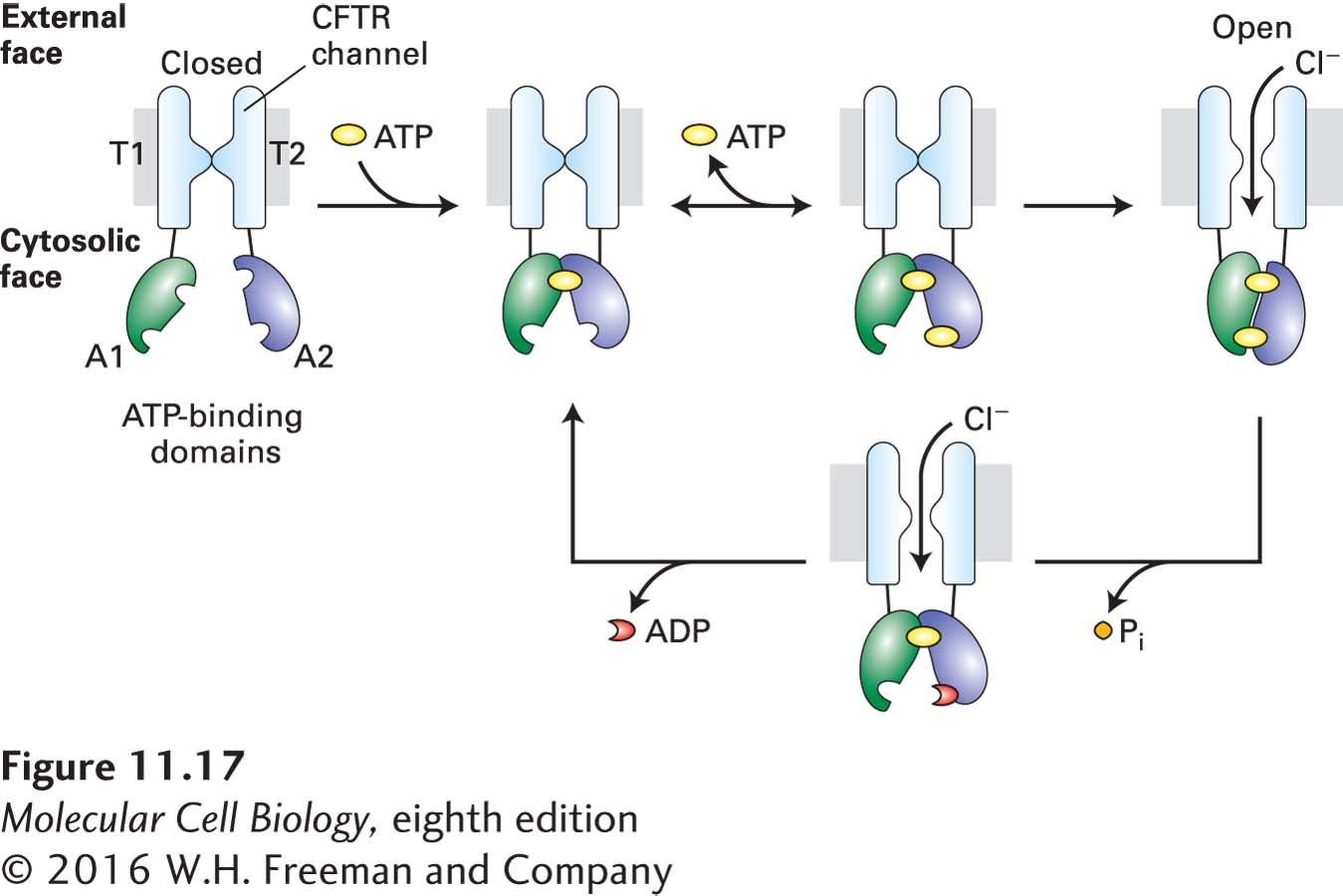
The Cl− channel of CFTR is normally closed. Channel opening is activated by phosphorylation of the R domain by a protein kinase (PKA, discussed in Chapter 15), which in turn is activated by an increase in cyclic AMP (cAMP), a small intracellular signaling molecule. Opening of the channel also requires sequential binding of two ATP molecules to the two A domains (Figure 11-17).
FIGURE 11-17Structure and function of the cystic fibrosis transmembrane regulator (CFTR). The regulatory (R) domain (not depicted) must be phosphorylated before ATP is able to power channel opening. Upon phosphorylation, one ATP (yellow circle) becomes tightly bound to the A1 domain (green). Binding of a second ATP to the A2 domain (blue) is followed by formation of a tight intramolecular A1–A2 heterodimer and slow channel opening. The relatively stable open state becomes destabilized by hydrolysis of the ATP bound at A2 to ADP (red crescent) and Pi. The ensuing disruption of the tight A1–A2 dimer interface leads to channel closure. T = transmembrane domain; A = cytosolic ATP-binding domain. See D. C. Gadsby et al., 2006, Nature440:477.
About two-thirds of all CF cases can be attributed to a single mutation in CFTR: deletion of Phe 508 in the ATP-binding A1 domain. At body temperature, the mutant protein fails to fold properly and to move to the cell surface, where it normally functions. Interestingly, if cells expressing the mutant protein are incubated at room temperature, the protein folds and accumulates normally on the plasma membrane, where it functions nearly as well as the wild-type CFTR channel. Recently a small molecule has been chemically synthesized that binds to this mutant CFTR protein in CF patients and stabilizes the folded form at 37 °C, allows it to traffic normally to the cell surface, and partially reverses the effects of the disease. Another CFTR mutation, Gly 551 to Asp, accounts for approximately 5 percent of CF cases and results in a channel that has normal surface expression but is defective in Cl− transport because the mutation disrupts ATP binding. Small molecules that increase the flow of Cl− ions through the mutant channel, called CFTR potentiators, are currently used to treat CF patients whose disease is caused by this mutation. These drugs represent some of the first successful personalized therapies that are based on a molecular understanding of the disease-causing protein.