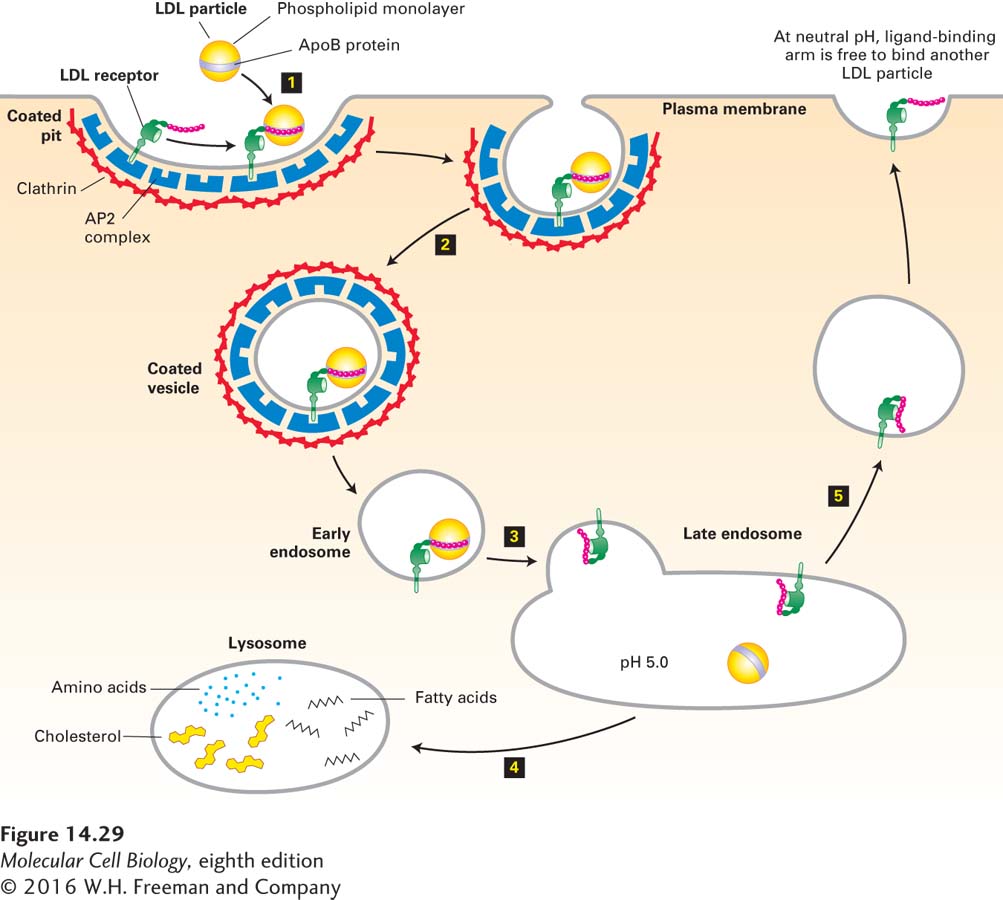
Receptors for Macromolecular Ligands Contain Sorting Signals That Target Them for Endocytosis
The key to understanding how LDL particles bind to the cell surface and are then taken up into endocytic vesicles was the discovery of the LDL receptor. The LDL receptor is an 839-
Page 661
![]() The discovery of the LDL receptor and an understanding of how it functions came from studying cells from patients with familial hypercholesterolemia (FH), a hereditary disease that is marked by elevated plasma LDL levels and is now known to be caused by mutations in the LDL receptor (LDLR) gene. In patients who have one normal and one defective copy of the LDLR gene (heterozygotes), LDL in the blood is increased about twofold. Those with two defective LDLR genes (homozygotes) have LDL levels that are from fourfold to sixfold higher than normal. Without medical intervention, FH heterozygotes commonly develop cardiovascular disease about 10 years earlier than normal people do, and FH homozygotes usually die of heart attacks before reaching their late twenties.
The discovery of the LDL receptor and an understanding of how it functions came from studying cells from patients with familial hypercholesterolemia (FH), a hereditary disease that is marked by elevated plasma LDL levels and is now known to be caused by mutations in the LDL receptor (LDLR) gene. In patients who have one normal and one defective copy of the LDLR gene (heterozygotes), LDL in the blood is increased about twofold. Those with two defective LDLR genes (homozygotes) have LDL levels that are from fourfold to sixfold higher than normal. Without medical intervention, FH heterozygotes commonly develop cardiovascular disease about 10 years earlier than normal people do, and FH homozygotes usually die of heart attacks before reaching their late twenties.
A variety of mutations in the LDLR gene can cause FH. Some mutations prevent the synthesis of the LDLR protein; others prevent proper folding of the receptor protein in the ER, leading to its premature degradation (see Chapter 13); and still other mutations reduce the ability of the LDL receptor to bind LDL tightly. A particularly informative group of mutant receptors are expressed on the cell surface and bind LDL normally but cannot mediate the internalization of bound LDL. In individuals with this type of defect, plasma-
A small number of individuals who exhibit the usual symptoms associated with FH produce normal LDL receptors. In these individuals, the gene encoding the AP2 subunit protein that binds the NPXY sorting signal is defective. As a result, LDL receptors are not incorporated into clathrin/AP2-
Mutational studies have shown that other cell-
In some cell-