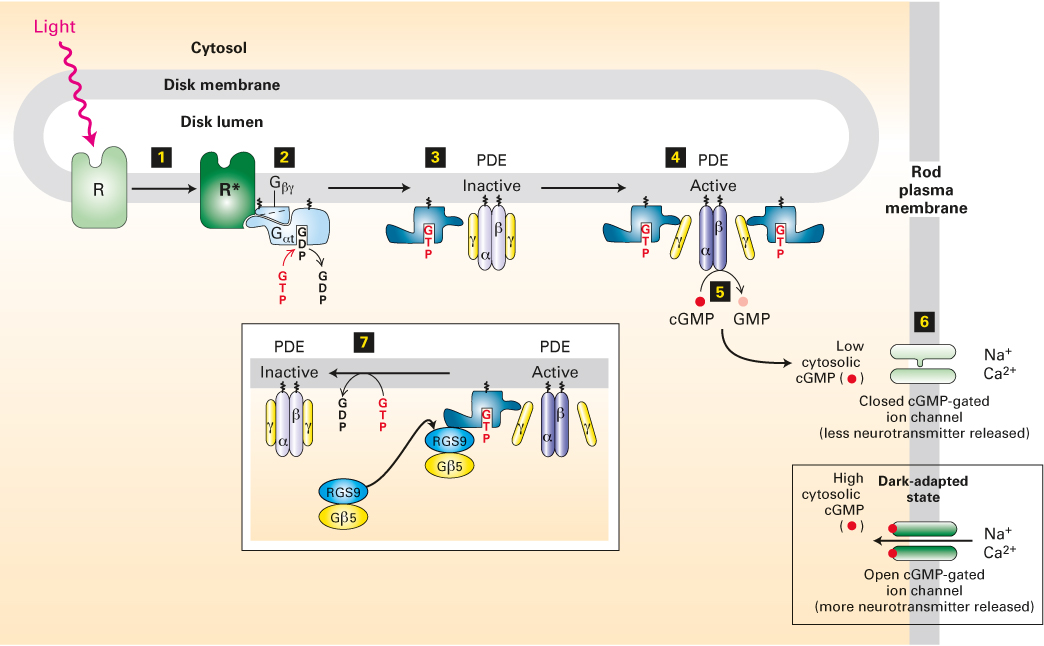
Activation of Rhodopsin by Light Leads to Closing of cGMP-Gated Cation Channels
In the dark, the membrane potential of a rod cell is about −30 mV, considerably more positive (less negative) than the resting potential (−60 to −90 mV) typical of neurons and other electrically active cells (see Chapter 11). This state of the membrane, called the depolarized state, causes rod cells in the dark to secrete neurotransmitters constantly, and thus the neurons with which they synapse are constantly being stimulated. The depolarized state of the plasma membrane of a resting rod cell is due to the presence of a large number of open nonselective ion channels that admit both Na+ and Ca2+ into the cell; recall from Chapter 11 that movement of cations (positively charged ions) such as Na+ and Ca2+ from the outside of the cell to the inside will reduce the magnitude of the inside-negative membrane potential, or depolarize the membrane. These nonselective cation channels open in response to (are “gated” by) binding of the second messenger cyclic guanosine monophosphate, or cGMP (see Figure 15-6). Rod-cell outer segments contain an unusually high concentration (~0.07 mM) of cGMP, which is continuously formed from GTP in a reaction catalyzed by guanylyl cyclase, and thus the cGMP-gated channels are mostly kept in the open state.
Absorption of light by rhodopsin leads to the closing of the nonselective cation channels, causing the membrane potential to become more negative inside. Unlike those associated with the muscarinic acetylcholine receptor discussed earlier, G proteins activated by rhodopsin do not act directly on ion channels. The closing of the cation channels in the rod-cell plasma membrane is caused by a marked reduction in the level of cGMP (Figure 15-20). Light absorption by rhodopsin induces rapid activation of a cGMP phosphodiesterase (PDE), which hydrolyzes cGMP to 5′-GMP; this light-induced drop in cGMP levels leads to channel closing. This, in turn, causes membrane hyperpolarization and a reduction in neurotransmitter release. The more photons absorbed by rhodopsin, the more cGMP is hydrolyzed, the more channels are closed, the fewer Na+ and Ca2+ ions cross the membrane from the outside, the more negative the membrane potential becomes, and the less neurotransmitter is released. The reduction in neurotransmitter release is transmitted to the brain by a series of neurons, where it is perceived as light.
FIGURE 15-20The light-activated rhodopsin pathway and the closing of cation channels in rod cells. In dark-adapted rod cells, a high level of cGMP keeps cGMP-gated nonselective cation channels open, leading to depolarization of the plasma membrane and neurotransmitter release. Light absorption generates activated rhodopsin, R* (step 1), which binds inactive, GDP-bound Gαt protein and mediates the exchange of GDP for GTP (step 2). The free Gαt·GTP generated then activates PDE by binding to its inhibitory γ subunits (step 3) and dissociating them from the catalytic α and β subunits (step 4). Relieved of their inhibition, the α and β subunits of PDE hydrolyze cGMP to GMP (step 5). The resulting decrease in cytosolic cGMP leads to dissociation of cGMP from the cation channels in the plasma membrane and the closing of those channels (step 6). The membrane then becomes transiently hyperpolarized, and neurotransmitter release is reduced. The complex of Gαt·GTP and the PDE γ subunits binds a GTPase-activating complex termed RGS9-Gβ5 (step 7); by hydrolyzing the bound GTP, this complex triggers the physiologically important rapid inactivation of the PDE. See V. Arshavsky and E. Pugh, 1998, Neuron20:11, and V. Arshavsky, 2002, Trends Neurosci.25:124.
As depicted in Figure 15-20, both Gαt·GTP and its effector, PDE, are localized via lipid anchors to the cytoplasmic face of the disk membranes of the rod cell. The Gαt·GTP complexes that are generated through activation of rhodopsin are able to move laterally along the membrane surface and bind to the two inhibitory γ subunits of PDE (see Figure 15-20; note that the stoichiometry is 1:1, i.e., one Gαt·GTP binds to one γ subunit). The binding of Gαt·GTP to the γ subunits releases the catalytically active αβ dimer, which then converts cGMP to GMP. This is a clear example of how signal-induced removal of an inhibitory subunit can quickly activate an enzyme, a common mechanism in signaling pathways.
Direct support for the role of cGMP in rod-cell activity has been obtained in patch-clamping studies using isolated patches of rod outer-segment plasma membrane, which contains abundant cGMP-gated cation channels. When cGMP is added to the cytosolic surface of these patches, there is a rapid increase in the number of open cation channels; cGMP binds directly to a site on the channel proteins to keep them open. Like the K+ channels discussed in Chapter 11, the cGMP-gated channel protein contains four subunits (see Figure 11-20). In this case, each of the subunits is able to bind a cGMP molecule. Three or four cGMP molecules must bind to each channel in order to open it; this cooperative allosteric interaction makes channel opening very sensitive to small changes in cGMP levels.
Page 696
Gαt contains an intrinsic GTPase activity that hydrolyzes a bound GTP to GDP, producing an inactive Gαt·GDP complex that can no longer associate with a PDE γ subunit. The intrinsic GTPase activity of Gαt in rod cells is accelerated by a specific GTPase-activating protein (GAP). In mammals, Gαt normally remains in the active GTP-containing state for only a fraction of a second. The rapid hydrolysis of GTP to GDP and subsequent inactivation of Gαt triggers release of the bound PDE γ subunits, which then rejoin the PDE α and β subunits to regenerate an inactive PDE αβγ2 tetramer. Thus PDE rapidly loses its activity, and the cGMP concentration begins to rise to pre-light-induction levels. Through this process, the eye can respond quickly to changing patterns of light associated with moving objects or as the direction of gaze changes.