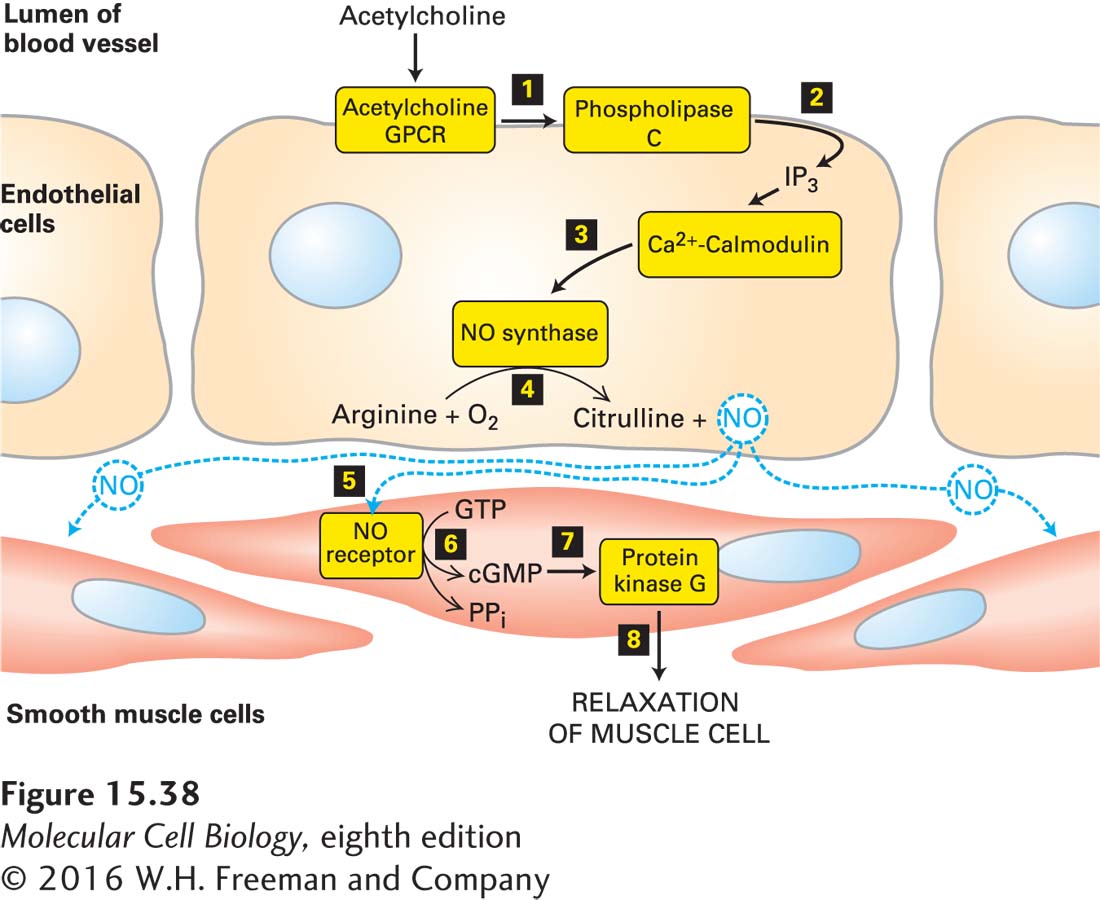
Signal-Induced Relaxation of Vascular Smooth Muscle Is Mediated by a Ca2+-Nitric Oxide-cGMP-Activated Protein Kinase G Pathway
In the late nineteenth century, Alfred Nobel (of the Nobel Prize) figured out how to improve nitroglycerine as an explosive that could be used in blasting rock and in mining, but for over a century nitroglycerin has also found an important use as a treatment for the intense chest pain of angina. It was known to slowly decompose in the body to nitric oxide (NO), which causes relaxation of the smooth muscle cells surrounding the blood vessels that “feed” the heart muscle itself, thereby increasing the diameter of the blood vessels and increasing the flow of oxygen-bearing blood to the heart muscle. One of the most intriguing discoveries in modern medicine is that NO, a toxic gas found in car exhaust, is in fact a natural signaling molecule.
Page 715
Definitive evidence for the role of NO in inducing relaxation of smooth muscle came from a set of experiments in which acetylcholine was added to experimental preparations of the smooth muscle cells that surround blood vessels (see Figure 1-25). Direct application of acetylcholine to these cells caused them to contract, as expected for vascular muscle cells. But addition of acetylcholine to the lumen of small isolated blood vessels caused the smooth muscles in those vessels to relax, not contract. Subsequent studies showed that in response to acetylcholine, the endothelial cells that line the lumen of a blood vessel were releasing some substance that in turn triggered muscle-cell relaxation. That substance turned out to be NO.
We now know that vascular endothelial cells contain a Gαo protein–coupled GPCR that binds acetylcholine and activates phospholipase C, leading to an increase in the level of cytosolic Ca2+. After Ca2+ binds to calmodulin, the Ca2+/calmodulin complex stimulates the activity of NO synthase, an enzyme that catalyzes formation of NO from O2 and the amino acid arginine. Because NO has a short half-life (2–30 seconds), it can diffuse only locally in tissues from its site of synthesis. In particular, NO diffuses from the vascular endothelial cell into neighboring smooth muscle cells, where it triggers muscle relaxation, which increases the diameter of the blood vessel (vasodilation) (Figure 15-38).
Figure 15-38The Ca2+/nitric oxide (NO)/cGMP pathway and the relaxation of vascular smooth muscle. Nitric oxide is synthesized in endothelial cells in response to activation of acetylcholine GPCRs, phospholipase C, and the subsequent elevation in cytosolic Ca2+ (steps 1–4). NO diffuses locally through tissues and activates an intracellular NO receptor with guanylyl cyclase activity in nearby smooth muscle cells 5. The resulting rise in cGMP 6 activates protein kinase G 7, leading to relaxation of the muscle and thus vasodilation 8. PPi = pyrophosphate. See C. S. Lowenstein et al., 1994, Ann. Intern. Med. 120:227, and H. K. Surks, 2007, Circ. Res.101:1078.
Page 716
The effect of NO on smooth muscle is mediated by the second messenger cGMP, which is formed by a cytosolic NO receptor expressed by smooth muscle cells. Binding of NO to the heme group in this receptor leads to a conformational change that increases its intrinsic guanylyl cyclase activity, leading to a rise in the cytosolic cGMP level. Most of the effects of cGMP are mediated by a cGMP-dependent protein kinase, also known as protein kinase G (PKG), that is regulated similarly to PKA except that the regulatory domain is part of the PKG polypeptide. This N-terminal domain contains a pseudosubstrate segment that binds to the kinase domain and inhibits its activity, as well as two cGMP-binding sites. Binding of cGMP induces a conformational change in the regulatory domain that prevents the pseudosubstrate from inhibiting the PKG kinase. In vascular smooth muscle, PKG activates a signaling pathway leading to relaxation of the cell and therefore dilation of the blood vessel. The dilation is caused in part by inhibition of Ca2+ channels in the endoplasmic reticulum (see Figure 15-34a) and a resulting decrease in cytosolic Ca2+ concentration; in Chapter 17 (page 808), we learn that in smooth muscle, a reduction in cytosolic Ca2+ causes a decrease in phosphorylation of a regulatory myosin light chain, disassembly of the actin-myosin contractile structure, and inhibition of muscle contraction. In vascular smooth muscle cells, cGMP acts indirectly via protein kinase G, in contrast to rod cells, in which cGMP acts directly by binding to and opening cation channels in the plasma membrane (see Figure 15-20).
Inhibitors of the cGMP-hydrolyzing enzyme PDE, which cause an elevation of cGMP in vascular smooth muscle cells, were originally developed as a treatment for male pattern baldness. A large clinical trial showed that they failed to increase hair growth, but a major side effect was noted—prolonged erections. Then several PDE inhibitors were quickly developed to treat erectile dysfunction. We’ll spare you the details here, but any interested reader can quickly find out how these widely used drugs function.