Somatic Cells Can Generate iPS Cells
Because of the inefficiency of somatic-
Several other transcription factors, and even certain small organic molecules, can replace the Oct4 gene in the Yamanaka reprogramming “cocktail.” Subsequent analysis led to the discovery that each of these factors directly activates transcription of the endogenous (cellular) Oct4 gene, leading to induction of pluripotency. Thus it was hypothesized that, over time, forced expression of transcription-
In fibroblasts, the chromatin of most pluripotency-
Page 984
![]() Because iPS cells can be derived from somatic cells of patients with difficult-
Because iPS cells can be derived from somatic cells of patients with difficult-
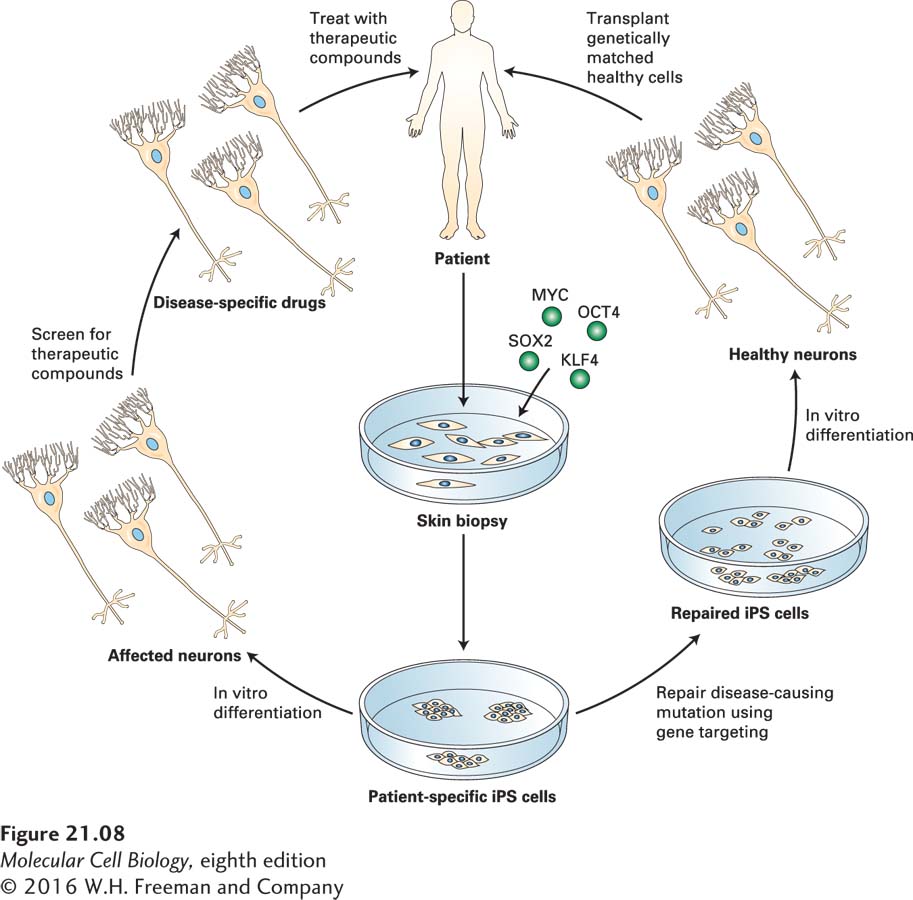
In approximately 10 percent of patients, the disease is dominantly inherited (familial ALS), but in 90 percent of patients, there is no apparent genetic linkage (sporadic ALS). An analysis of the underlying causes of the disease at a molecular and cellular level was impossible for many years because one cannot simply extract neurons or the surrounding glial cells from living humans and analyze or culture them.
In about 20 percent of patients with familial ALS, there is a point mutation in the gene SOD1, encoding Cu/Zn superoxide dismutase 1; the mutant SOD1 protein forms aggregates that can damage cells. About 40 percent of patients with familial ALS and 10 percent of patients with the noninherited form have a mutation in the C9ORF72 gene (of unknown function; called chromosome 9 open reading frame 72). This mutation also often occurs in people with frontotemporal dementia, the second most common form of dementia after Alzheimer’s disease, explaining why some people develop both diseases simultaneously. The mRNA transcribed from normal human C9ORF72 genes has up to 30 repeats of the hexanucleotide GGGGCC, but mutant ALS-
Page 985
In several studies, iPS cells derived from the skin cells of elderly patients with these and other familial and sporadic forms of the disease were successfully differentiated in culture to form motor neurons; this success demonstrated the feasibility of leveraging the self-
In a separate study to dissect the molecular cause of ALS, motor neurons were generated from human ES or iPS cells and cultured with primary human astrocytes, a type of glial cell that surrounds neurons and regulates several of their functions (see Figure 22-17). Many of the motor neurons died if the astrocytes expressed the mutant form of SOD1, but not if they expressed the wild-
Page 986
In these and several other studies, researchers screened thousands of small organic molecules, including many approved as drugs for treatment of other unrelated diseases, for those that could reverse the abnormalities in the ALS iPS cell–