The Pro-apoptotic Proteins Bax and Bak Form Pores and Holes in the Outer Mitochondrial Membrane
In vertebrate cells, Bax or Bak is required for mitochondrial damage and induction of apoptosis. These two similar pro-apoptotic proteins contain three of the BH1–4 domains (see Figure 21-39) and have three-dimensional structures very similar to that of the anti-apoptotic members of the family. As evidence for the role of these proteins in promoting apoptosis, most mice lacking both Bax and Bak die in utero, and those that survive show significant developmental defects, including the persistence of interdigital webs and accumulation of extra cells in the central nervous and hematopoietic systems. Cells isolated from these mice are resistant to virtually all apoptotic stimuli. Conversely, overproduction of Bax in cultured cells induces apoptotic death.
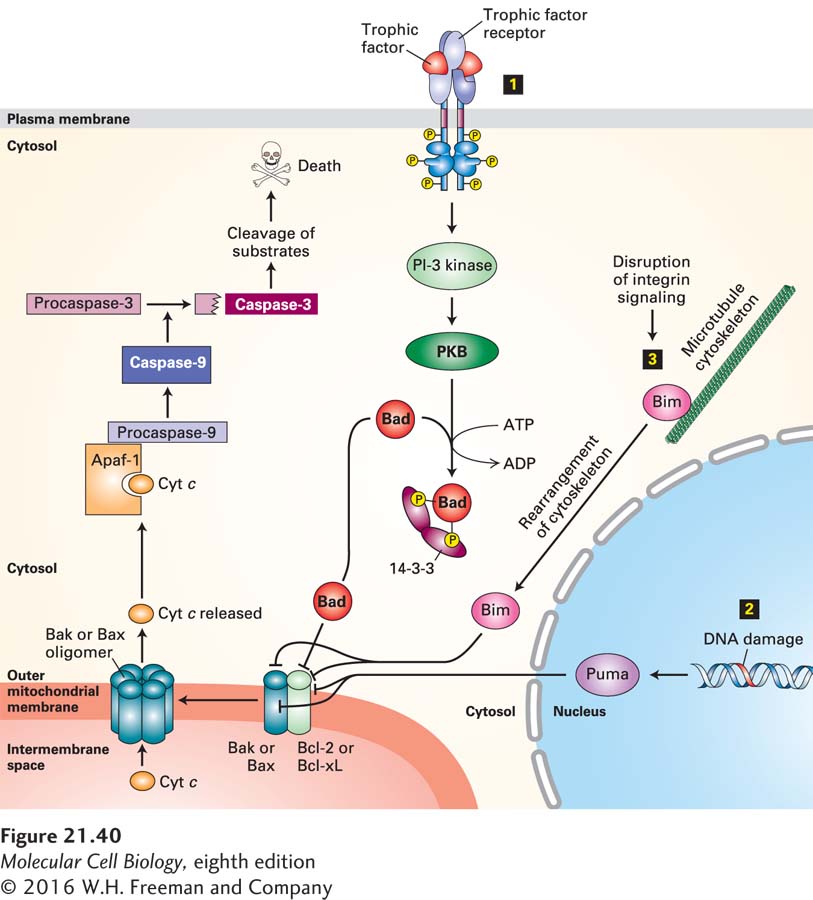
Bak resides in the outer mitochondrial membrane, normally tightly bound to Bcl-2 or the related protein Bcl-xL (Figure 21-40). When released from Bcl-2—either by being present in excess, by being displaced by the binding of certain BH3-only proteins to Bcl-2, or by binding directly to other BH3-only proteins—Bak forms oligomers that generate pores in the outer mitochondrial membrane. Bax is mainly cytosolic, with a small fraction attached to mitochondria; binding of certain pro-apoptotic proteins, discussed later, causes Bax, like Bak, to oligomerize and insert into the outer mitochondrial membrane, forming pores.
FIGURE 21-40Integration of multiple signaling pathways in vertebrate cells that regulate outer mitochondrial membrane permeability and apoptosis. In healthy cells, the anti-apoptotic protein Bcl-2, or its homolog Bcl-xL, binds to Bak or Bax pro-apoptotic proteins, blocking the ability of Bak or Bax to oligomerize and form pores in the outer mitochondrial membrane. Binding of any of several BH3-only proteins, including Bad, Bim, and Puma, to Bcl-2 or directly to Bak or Bax causes Bak or Bax to dissociate from Bcl-2 and form oligomeric pores and holes in the outer mitochondrial membrane. These holes allow cytochrome c to enter the cytosol, where it binds to the adapter protein Apaf-1, promoting caspase activation that initiates the apoptotic cascade and leads to cell death. Several stimuli trigger or repress this apoptotic pathway. Step 1 The presence of specific trophic factors (e.g., NGF) leads to activation of their cognate receptor tyrosine kinases (e.g., TrkA) and activation of the PI-3 kinase–PKB (protein kinase B) pathway (see Figure 16-29). PKB phosphorylates Bad, and phosphorylated Bad then forms a complex with a cytosolic 14-3-3 protein. This sequestered Bad is unable to bind to Bcl-2. In the absence of trophic factors, nonphosphorylated Bad binds to Bcl-2, releasing Bax and Bak and allowing them to form oligomeric membrane pores and holes. Step 2 DNA damage or ultraviolet irradiation leads to induction of synthesis of the BH3-only Puma protein. Puma binds to Bak and Bax as well as to Bcl-2, allowing Bak and Bax to form oligomeric pores. Step 3 Removal of a cell from its substratum disrupts integrin signaling, leading to release of the BH3-only Bim protein from the cytoskeleton. Bim also binds to Bak and Bax to promote pore formation. See D. Ren et al., 2010, Science330:1390 and Czabootar et al., 2014, Nat. Rev. Mol. Cell. Biol.15:49.
Recall from Chapter 12 that mitochondria are constantly undergoing fusion as well as fission (the latter when two daughter mitochondria separate from each other). Both Bak and Bax, when oligomerized, accumulate at sites of mitochondrial fission, causing holes to form in the outer membrane at those sites. Both pores and holes in the outer mitochondrial membrane allow release into the cytosol of mitochondrial proteins such as cytochrome c that, in normal healthy cells, are localized to the intermembrane space.
As depicted in Figure 21-35, released cytochrome c activates caspase-9—in part by binding to and activating Apaf-1 and in part through as yet unknown mechanisms. As evidence for this regulatory pathway, overproduction of Bcl-2 in cultured cells blocks release of cytochrome c and blocks apoptosis; conversely, overproduction of Bax promotes release of cytochrome c into the cytosol and promotes apoptosis. Moreover, injection of cytochrome c directly into the cytosol of cells induces apoptosis.