Opening of Acetylcholine-Gated Cation Channels Leads to Muscle Contraction
In this section we look at how binding of neurotransmitters by receptors on postsynaptic cells leads to changes in the cells’ membrane potential, using the communication between motor neurons and muscles as an example. At these synapses, called neuromuscular junctions, acetylcholine is the neurotransmitter. A single axon terminus of a frog motor neuron may contain a million or more synaptic vesicles, each containing 1000–10,000 molecules of acetylcholine; these vesicles often accumulate in rows in the active zone (see Figures 22-23 and 22-24). Such a neuron can form synapses with a single skeletal muscle cell at several hundred points.
The nicotinic acetylcholine receptor, which is expressed in muscle cells, is a ligand-gated channel that admits both K+ and Na+. These receptors are also produced in brain neurons and are important in learning and memory; loss of these receptors is observed in schizophrenia, epilepsy, drug addiction, and Alzheimer’s disease. Antibodies against acetylcholine receptors constitute a major part of the autoimmune reactivity in the disease myasthenia gravis. The receptor is so named because it is bound by nicotine; it has been implicated in nicotine addiction in tobacco smokers. There are at least 14 different isoforms of the receptor, which assemble into homo- and heteropentamers with varied properties. Given their many physiological functions and their role in disease, these various isoforms are important targets for new drug development
The effect of acetylcholine on this receptor can be determined by patch-clamp recording from isolated outside-out patches of muscle plasma membranes. Outside-out patch-clamp recording is a technique that measures the effects of extracellular solutes on channel receptors within the isolated patch (see Figure 11-22c). Such measurements have shown that acetylcholine causes opening of a cation channel in the receptor capable of transmitting 15,000–30,000 Na+ and K+ ions per millisecond. However, since the resting potential of the muscle plasma membrane is near EK, the potassium equilibrium potential, opening of acetylcholine receptor channels causes little increase in the efflux of K+ ions; Na+ ions, on the other hand, flow into the muscle cell, driven by the Na+ electrochemical gradient.
Page 1058
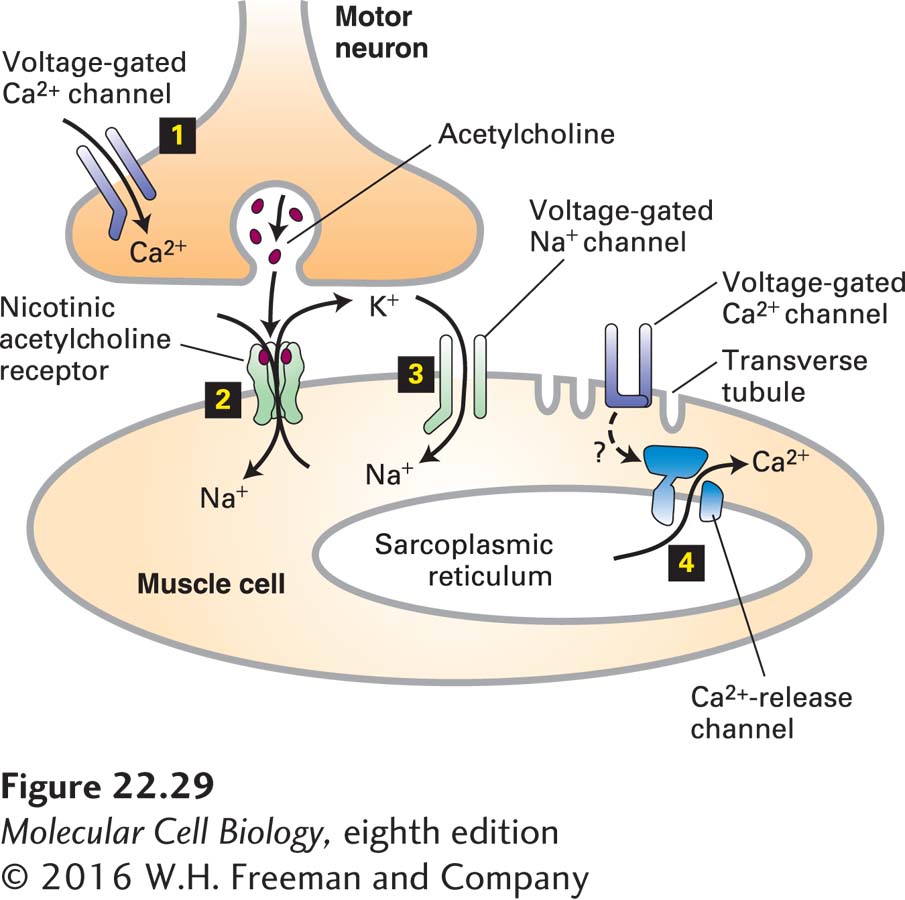
The simultaneous increase in permeability to Na+ and K+ ions following binding of acetylcholine produces a net depolarization to about −15 mV from the muscle resting potential of −85 to −90 mV. As shown in Figure 22-29, this localized depolarization of the muscle plasma membrane triggers opening of voltage-gated Na+ channels, leading to generation and conduction of an action potential in the muscle cell surface membrane by the same mechanisms described previously for neurons. When the membrane depolarization reaches transverse tubules (see Figure 17-33), specialized invaginations of the plasma membrane, it acts on Ca2+ channels in the plasma membrane apparently without causing them to open. This in turn triggers the opening of adjacent Ca2+-release channels in the sarcoplasmic reticulum membrane. The subsequent flow of stored Ca2+ ions from the sarcoplasmic reticulum into the cytosol raises the cytosolic Ca2+ concentration sufficiently to induce muscle contraction.
FIGURE 22-29Sequential activation of gated ion channels at a neuromuscular junction. Arrival of an action potential at the terminus of a presynaptic motor neuron induces opening of voltage-gated Ca2+ channels in the neuron (step 1) and subsequent release of acetylcholine, which triggers opening of the ligand-gated acetylcholine receptors in the muscle plasma membrane (step 2). The open receptor channel allows an influx of Na+ and an efflux of K+ from the muscle cell. The Na+ influx produces a localized depolarization of the membrane, leading to opening of voltage-gated Na+ channels and generation of an action potential (step 3). When the spreading depolarization reaches transverse tubules, it is sensed by voltage-gated Ca2+ channels in the plasma membrane. Through an unknown mechanism (indicated as ?) these channels remain closed but influence Ca2+ channels in the sarcoplasmic reticulum membrane (a network of membrane-bound compartments in muscle), which release stored Ca2+ into the cytosol (step 4). The resulting rise in cytosolic Ca2+ causes muscle contraction by mechanisms discussed in Chapter 17.
Careful monitoring of the membrane potential of the muscle membrane at a synapse with a cholinergic motor neuron has demonstrated spontaneous, intermittent, and random ~2-ms depolarizations of about 0.5–1.0 mV in the absence of stimulation of the motor neuron. Each of these depolarizations is caused by the spontaneous release of acetylcholine from a single synaptic vesicle in the neuron. Indeed, demonstration of such spontaneous small depolarizations led to the notion of the quantal release of acetylcholine (later applied to other neurotransmitters) and thereby led to the hypothesis of vesicle exocytosis at synapses. The release of one acetylcholine-containing synaptic vesicle results in the opening of about 3000 ion channels in the postsynaptic membrane, far short of the number needed to reach the threshold depolarization that induces an action potential. Clearly stimulation of muscle contraction by a motor neuron requires the nearly simultaneous release of acetylcholine from numerous synaptic vesicles.