Inherited Mutations in Tumor-Suppressor Genes Increase Cancer Risk
Individuals with inherited mutations in tumor-suppressor genes have a hereditary predisposition to certain cancers. Such individuals generally inherit a germ-line mutation in one allele of the gene; somatic mutation of the second allele facilitates tumor progression. A classic case is retinoblastoma, which is caused by loss of function of RB, the first tumor-suppressor gene to be identified. As discussed in Chapter 19, the protein encoded by RB regulates cell cycle entry.
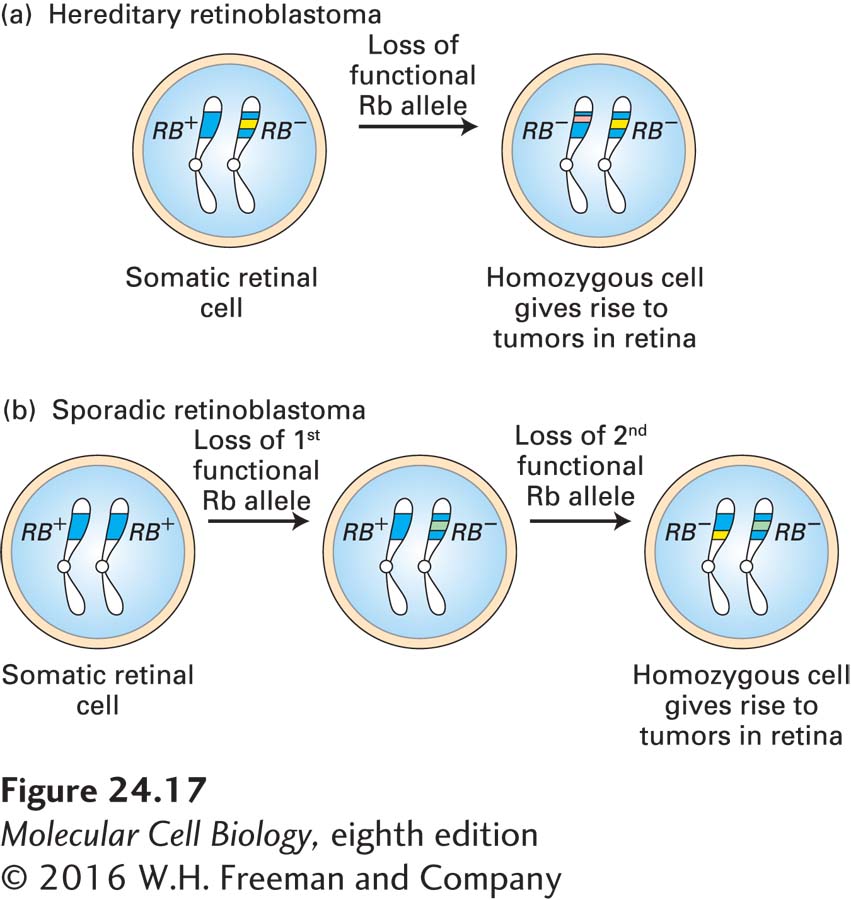
Children with hereditary retinoblastoma inherit one defective copy of the RB gene, sometimes seen as a small deletion on one of the two copies of chromosome 13. These children develop multiple retinal tumors early in life and generally in both eyes. The loss or inactivation of the normal RB gene on the other chromosome is an essential step in tumor formation, giving rise to a cell that produces no functional Rb protein (Figure 24-17a). Individuals with sporadic retinoblastoma, in contrast, inherit two normal RB alleles, each of which has undergone a loss-of-function somatic mutation or loss in a single retinal cell (Figure 24-17b). Because losing two copies of the RB gene is far less likely than losing one, sporadic retinoblastoma is rare and usually affects only one eye.
FIGURE 24-17Role of spontaneous somatic mutation in retinoblastoma. This disease is marked by retinal tumors that arise from cells carrying two mutant RB− alleles. (a) In hereditary (familial) retinoblastoma, a child inherits a normal RB+ allele from one parent and a mutant RB− allele from the other parent. When the second normal allele is lost in a heterozygous somatic retinal cell, a cell is generated that lacks any Rb gene function. (b) In sporadic retinoblastoma, a child inherits two normal RB+ alleles. Two separate Rb loss events must occur in a particular retinal cell to produce a cell lacking all Rb function.
If retinal tumors are removed before they become malignant, children with hereditary retinoblastoma often survive until adulthood and produce children, but are at an increased risk of developing other types of tumors later in life. Because their germ cells contain one normal and one mutant RB allele, these individuals will, on average, pass on the mutant allele to half their children and the normal allele to the other half. Children who inherit the normal allele are normal if their other parent has two normal RB alleles. However, those who inherit the mutant allele have the same enhanced predisposition to develop retinal tumors as their affected parent, even though they inherit a normal RB allele from their other, normal parent. Thus the tendency to develop retinoblastoma is inherited as a dominant trait: one mutant copy is sufficient to predispose a person to develop the cancer.
As we will see shortly, many human tumors (not just retinal tumors) contain mutant RB alleles or mutations affecting other components of the Rb pathway; most of these tumors arise as the result of somatic mutations. Although hereditary retinoblastoma cases number about 100 per year in the United States, about 100,000 other cancer cases each year involve RB mutations acquired postconception.
Similar hereditary predispositions to other cancers have been associated with inherited mutations in other tumor-suppressor genes. For example, individuals who inherit a germ-line mutation in one APC allele develop thousands of precancerous intestinal polyps (see Figure 24-12). Since there is a high probability that one or more of these polyps will progress to malignancy, such individuals have a greatly increased risk of developing colon cancer before the age of 50. Screening for polyps by colonoscopy is a good idea for people 50 or older, even when no APC mutation is known to be present. Likewise, women who inherit one mutant allele of BRCA1, another tumor-suppressor gene, have a 60 percent probability of developing breast cancer by age 50, whereas those who inherit two normal BRCA1 alleles have a 2 percent probability of doing so. Heterozygous BRCA1 mutations also increase the lifetime risk of ovarian cancer from 2 percent to 15–40 percent. The BRCA1 protein is involved in repairing radiation-induced DNA damage. In women who inherit one mutant BRCA1 allele, loss of the second BRCA1 allele, together with other mutations, is required for a normal mammary duct cell to become malignant. However, BRCA1 generally is not mutated in sporadic breast cancer.
Page 1154
Estimates vary, but hereditary cancers (cancers that arise due in part to an inherited version of a gene) are thought to constitute about 10 percent of human cancers. Further work tracing the contributions of human genes seems likely to increase the percentage. It is important to remember, however, that the inherited germ-line mutation alone is not sufficient to cause tumor development. Not only must the inherited normal tumor-suppressor allele be lost or inactivated, but mutations affecting other genes must also occur for cancer to develop. Thus a person with a recessive tumor-suppressor gene mutation can be exceptionally susceptible to environmental mutagens such as radiation.
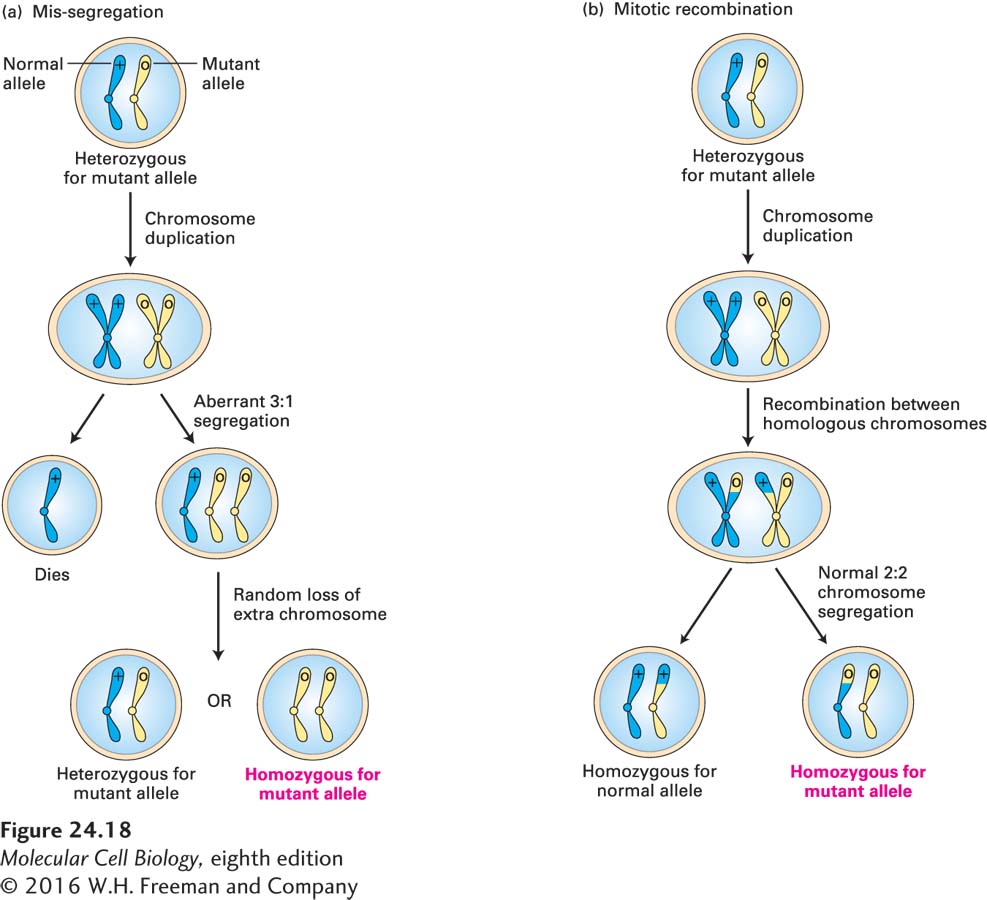
Mutation in only one copy of a tumor-suppressor gene itself typically does not cause cancer because the remaining normal allele prevents aberrant growth. However, the subsequent loss or inactivation of the remaining normal allele in a somatic cell, referred to as loss of heterozygosity (LOH) causes cancer to develop. Three mechanisms exist that can cause the loss of the normal allele. First, the normal allele can become inactive due to a de novo inactivating mutation or deletion. Second, chromosome mis-segregation, as outlined in Figure 24-18a, can cause loss of the chromosome carrying the normal allele. Neither mechanism is particularly frequent. By far the most frequent mechanism for LOH is mitotic recombination between a chromatid bearing the normal allele and a homologous chromatid bearing a mutant allele. As illustrated in Figure 24-18b, subsequent chromosome segregation can generate a daughter cell that is homozygous for the mutant tumor-suppressor allele.
Page 1155
FIGURE 24-18Two mechanisms for loss of heterozygosity (LOH) of tumor-suppressor genes. A cell containing one normal and one mutant allele of a tumor-suppressor gene is generally phenotypically normal. (a) If formation of the mitotic spindle is defective, then the duplicated chromosomes bearing the normal and mutant alleles may segregate in an aberrant 3:1 ratio. A daughter cell that receives three chromosomes of a type can lose one, restoring the normal 2n chromosome number. Sometimes the resultant cell will contain one normal and one mutant allele, but sometimes it will be homozygous for the mutant allele. Such aneuploidy (abnormal chromosome constitution) is generally damaging or lethal to relatively undifferentiated cells that have to develop into the many complex structures of an organism, but can often be tolerated in clones of cells that have limited fates and duties. (b) Mitotic recombination between a chromosome with a wild-type and a mutant allele, followed by chromosome segregation, can produce a cell that contains two copies of the mutant allele.