Mutations That Promote Unregulated Passage from G1 to S Phase Are Oncogenic
Once a cell progresses past a certain point in G1, called the restriction point, it becomes irreversibly committed to entering S phase and replicating its DNA (see Figure 19-12). Cyclin Ds, cyclin-dependent kinases (CDKs), and the Rb protein are all elements of the control system that regulates passage through the restriction point.
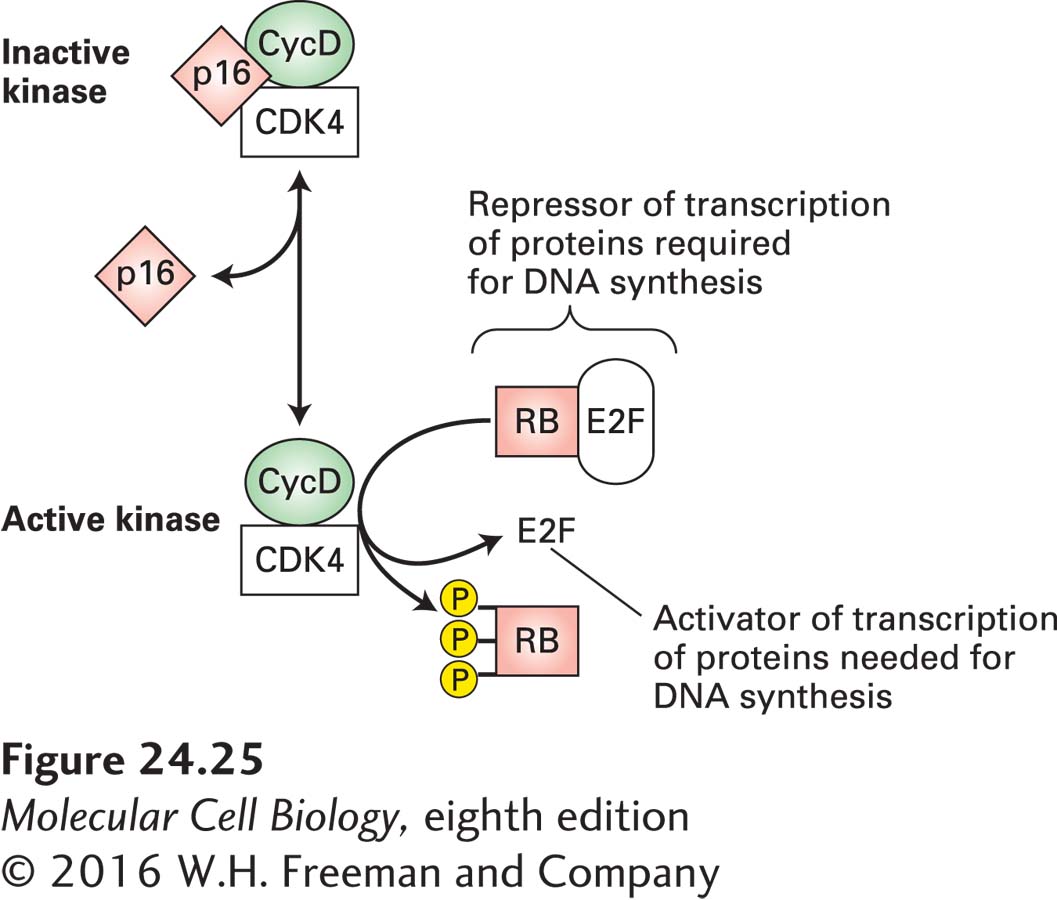
The pathway that controls entry into the cell cycle is estimated to be misregulated in approximately 80 percent of human cancers. At the heart of this pathway are cyclin D-CDK4/6 complexes and the transcription inhibitor RB (Figure 24-25). The expression of cyclin D genes is induced by many extracellular growth factors, or mitogens. These cyclins assemble with a partner, CDK4 or CDK6, to generate catalytically active cyclin-CDK complexes, whose kinase activity promotes progression through G1. Mitogen withdrawal prior to passage through the restriction point leads to accumulation of two CDK inhibitors. As described in Chapter 19, these two proteins, p15 and p16, bind to cyclin D–CDK4/6 complexes and inhibit their activity, thereby causing G1 arrest. The transcription inhibitor RB is controlled by cyclin D–CDK4/6 phosphorylation. Nonphosphorylated RB binds to E2F transcription factors, which stimulate transcription of genes encoding proteins required for DNA synthesis. Under normal circumstances, phosphorylation of RB protein is initiated midway through G1 by active cyclin D–CDK4/6 complexes. RB phosphorylation is completed by cyclin E–CDK2 complexes in late G1, allowing release and activation of E2Fs and progression from G1 to S. The complete phosphorylation of RB and its disassociation from E2Fs irreversibly commits the cell to DNA synthesis.
FIGURE 24-25Restriction point control. Nonphosphorylated RB protein binds transcription factors collectively called E2Fs and thereby prevents E2F-mediated transcriptional activation of many genes whose products (e.g., DNA polymerase) are required for DNA synthesis. The kinase activity of cyclin D–CDK4/6 phosphorylates RB, thereby inactivating RB and activating E2Fs; cyclin D–CDK4/6 activity is inhibited by p16. Overproduction of cyclin D, a positive regulator, or loss of the negative regulators p16 and RB commonly occurs in human cancers.
Most tumors contain an oncogenic mutation that causes the overproduction or loss of one of the components of the pathway that controls entry into S phase, so that the cells are propelled into S phase in the absence of the proper extracellular growth signals. For example, elevated levels of cyclin D1, one of the three cyclin Ds, are found in many human cancers. In certain tumors of antibody-producing B lymphocytes, the cyclin D1 gene is translocated such that its transcription is under the control of an antibody-gene enhancer, causing elevated cyclin D1 production throughout the cell cycle irrespective of extracellular signals. (This phenomenon is analogous to the MYC translocation in Burkitt’s lymphoma cells discussed earlier.) That cyclin D1 can function as an oncoprotein was shown by studies with transgenic mice in which the cyclin D1 gene was placed under the control of an enhancer specific for mammary duct cells. Initially, the duct cells underwent hyperproliferation, and eventually breast tumors developed in these transgenic mice. A second mechanism that can lead to overproduction of cyclin D is gene amplification. Amplification of the cyclin D1 gene and concomitant overproduction of the cyclin D1 protein is common in human breast cancers; the extra cyclin D1 helps to drive cells through the cell cycle.
We have already seen that inactivating mutations in both RB alleles lead to childhood retinoblastoma, a relatively rare type of cancer. However, loss of RB gene function is also found in more common cancers that arise later in life (e.g., carcinomas of lung, breast, and bladder). These tissues, unlike retinal tissue, probably produce other proteins (e.g., p107 and p130, both structurally related to RB) whose function is redundant with that of RB, and thus RB is not so critical for preventing cancer in these tissues. In the retina, however, regulation of cell cycle entry appears to rely exclusively on the RB protein, which is why patients heterozygous for the RB gene first develop tumors in this tissue. RB function can be eliminated not only by inactivating mutations, but also by the binding of an inhibitory protein, designated E7, that is encoded by human papillomavirus (HPV), another nasty viral trick to create virus-producing tissue. At present, this binding is known to occur only in cervical and oropharyngeal cancers.
The proteins that function as cyclin-CDK inhibitors play an important role in regulating the cell cycle. In particular, loss-of-function mutations that prevent p16 from inhibiting cyclin D–CDK4/6 kinase activity are common in several human cancers. As Figure 24-25 makes clear, loss of p16 mimics overproduction of cyclin Ds. Thus p16 normally acts as a tumor suppressor. Although the p16 tumor-suppressor gene is deleted in some human cancers, the p16 sequence is normal in others. In some of these latter cancers (e.g., lung cancer), the p16 gene, or genes encoding other functionally related proteins, is inactivated by hypermethylation of its promoter region, which prevents its transcription. What promotes this change in the methylation of p16 is not known, but it prevents production of this important cell cycle control protein.
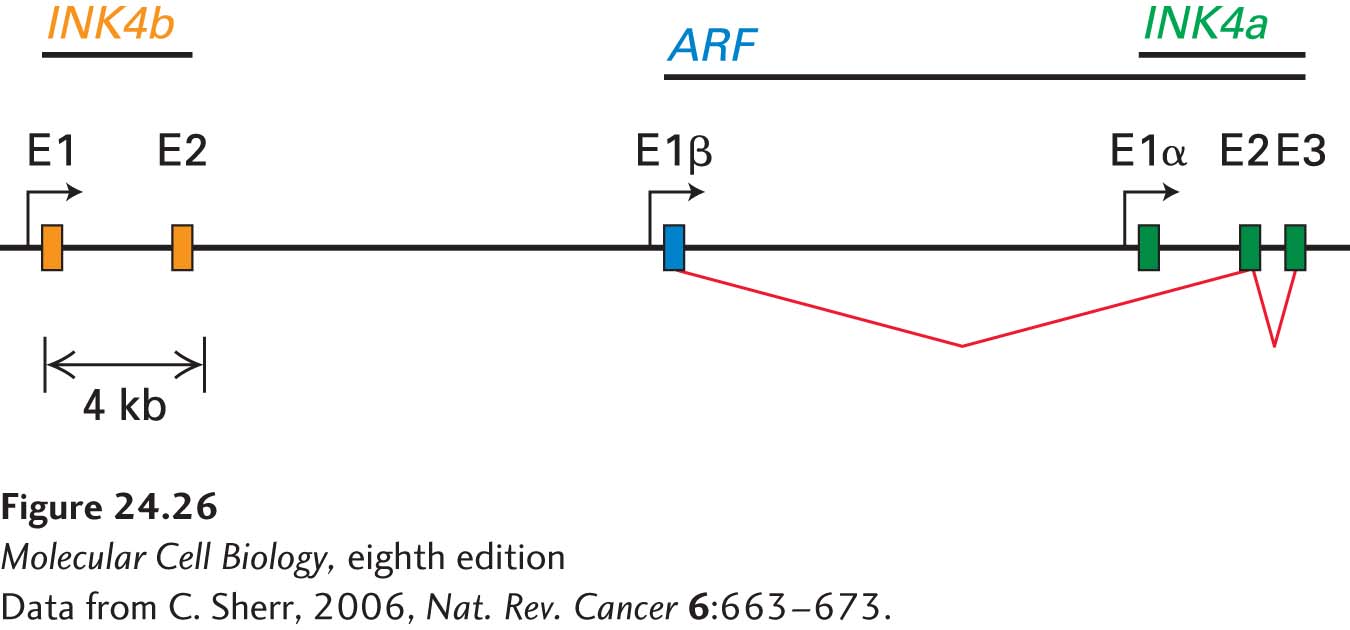
The locus encoding p16 is highly unusual in that it encodes no less than three tumor-suppressor genes, which makes it the most vulnerable locus in the human genome to oncogenic changes. In addition to harboring the p16-encoding gene, INK4a (CDKN2A), it has the INK4b (CDKN2B) locus immediately upstream, which encodes p15, another cyclin D–CDK4/6 inhibitor (Figure 24-26). In addition, the locus encodes a key activator of the tumor suppressor p53. This protein, p14ARF (p19ARF in the mouse), is encoded by an exon upstream of the first INK4a exon and shares its exon 2 and exon 3 with INK4a. As we will see next, this protein controls the stability of p53. Thus mutations in this locus can simultaneously affect the two major tumor-suppressor pathways in the cell, the RB and p53 pathways.
Page 1165
[Data from C. Sherr, 2006, Nat. Rev. Cancer6:663–673.]
FIGURE 24-26The INK4b-ARF-INK4a locus encodes three tumor-suppressor genes. Exons are designated as E. The two INK4b exons (orange) are located upstream of the ARF/INK4a locus. ARF (blue) is encoded by a unique E1β exon but shares exons E2 and E3 with INK4a (green). INK4b and INK4a encode p15 and p16, respectively. ARF encodes a p53 activator.
[Data from C. Sherr, 2006, Nat. Rev. Cancer6:663–673.]