Abnormally Folded Proteins Can Form Amyloids That Are Implicated in Diseases
After it is synthesized, a protein may fold into an alternative, abnormal three-dimensional structure as the result of mutations, inappropriate covalent modifications, or chemical (e.g., pH) or physical (e.g., heat) alterations in its environment. Misfolding or denaturation can lead to a loss of the normal function of the protein and can result in the protein being marked for destruction (proteolytic degradation), as described later in this chapter. However, when degradation is incomplete or fails to keep pace with the production of misfolded protein, the misfolded protein or its proteolytic fragments can accumulate either inside or outside of cells in aggregates, or plaques, in various organs, including joints between bones, the liver, and the brain. Even those proteins or protein fragments that are normally highly resistant to aggregation, as is the case for intrinsically disordered proteins or protein fragments, will form aggregates if their concentrations are sufficiently elevated or when there are changes in environmental conditions. As noted above, such aggregates can either be amorphous or have a well-organized structure, which most commonly is the amyloid state. Strikingly, many diverse proteins can each aggregate into amyloid fibrils that have a common structure, called a cross-β sheet (Figure 3-20a). Short segments, generally 6–12 residues long, in the unfolded or misfolded proteins hydrogen-bond to each other, forming a long array, or filament, of β sheets. In these arrays, each β strand is nearly perpendicular to the long axis of the filament, and two long, nearly flat β sheets pack closely together and twist around each other to form protofilaments, which then assemble together into thicker filaments, called amyloid fibrils. Within each protofilament the β strands can be either parallel or antiparallel (see Figure 3-5). Although some proteins form amyloid fibrils in their native, functional states, most amyloids are considered to be consequences of protein misfolding.
[Part (b, left) republished with permission of Elsevier, from Dobson, C.M., “Protein misfolding, evolution and disease,” Trends in Biochemical Science 1999, 24(9):329-332. Fig. 3. Part (b, right) reprinted by permission from Macmillan Publishers Ltd: from Knowles et al., Nat. Rev. Mol. Cell Biol. 2014, 15(6):384-396. Fig. 3a. Part (c) Thomas Deerinck, NCMIR/Science Source.]
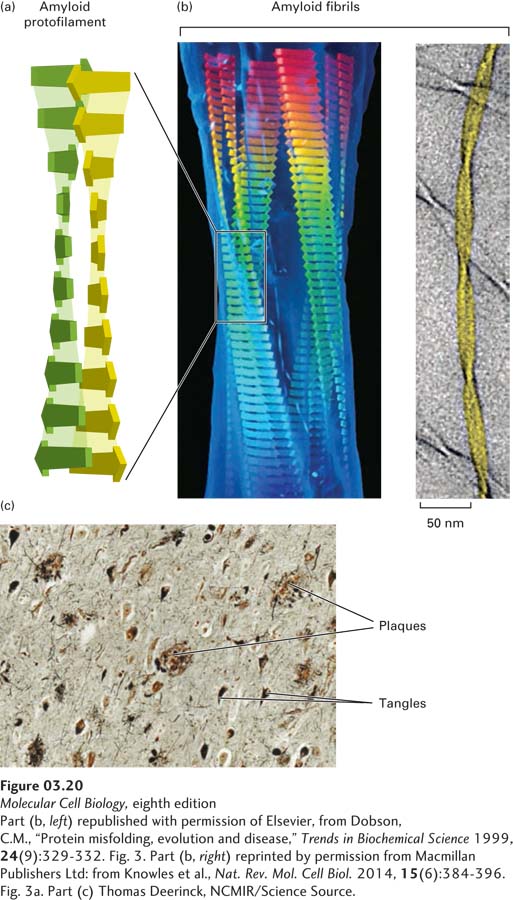
FIGURE 3-20Misfolded proteins can form ordered amyloid aggregates based on a cross-β sheet structure. (a) In unfolded segments of proteins and polypeptides, exposed segments 6–12 residues long (short flat arrows) can assemble into β sheets (see also Figure 3-5) in which each β strand is oriented nearly perpendicularly to the long axis (vertical in this figure) of the resultant amyloid protofilament and hydrogen-bonded (light shading) to the strands above and below. Two long, nearly flat sheets pack closely together and twist around each other to form amyloid protofilaments, which then assemble together into thicker filaments called amyloid fibrils (b). Amyloid fibrils can be composed of varying numbers of protofilaments. A model of a four-protofilament-containing fibril fit into the electron density of acid-denatured insulin fibrils (left) and a cryoelectron microscopic image of two-protofilament-containing fibrils of fragments of transthyretin with an NMR-based model (yellow). Fibrils can aggregate into macroscopic plaques and tangles that are deposited in tissues and, when stained, are large enough to be visible using light microscopy. (c) Microscopic view of a section of human brain tissue from a patient with Alzheimer’s disease with multiple amyloid plaques and fibrillary tangles.
[Part (b, left) republished with permission of Elsevier, from Dobson, C.M., “Protein misfolding, evolution and disease,” Trends in Biochemical Science 1999, 24(9):329-332. Fig. 3. Part (b, right) reprinted by permission from Macmillan Publishers Ltd: from Knowles et al., Nat. Rev. Mol. Cell Biol. 2014, 15(6):384-396. Fig. 3a. Part (c) Thomas Deerinck, NCMIR/Science Source.]
Amyloids were first recognized in protein aggregates that are deposited in tissues, are resistant to enzymatic degradation, and are associated with dozens of diseases, called amyloidoses. These diseases include neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease in humans and transmissible spongiform encephalopathy (“mad cow” disease) in cows and sheep. Each of these diseases is characterized by the presence of filamentous plaques in a deteriorating brain (Figure 3-20b). Amyloidoses most commonly occur with aging; however, mutations in the genes encoding the aggregating protein can result in early amyloid formation and disease onset. The amyloid fibrils composing the plaques derive from abundant natural proteins. For example, fragments of the amyloid precursor protein, which is embedded in the plasma membrane, form the plaque found in the brains of patients with Alzheimer’s disease; and prion protein, an “infectious” protein, forms fibrils in prion diseases. In Alzheimer’s disease, a hyperphosphorylated form of the protein tau, normally a microtubule-binding protein (see Chapter 18), forms twisted fibers called “tangles.” These amyloids, either as relatively short, water-soluble protofilaments or as long, insoluble fibrils, are thought to be toxic and to contribute directly to the pathology of amyloidoses.