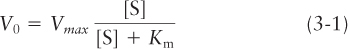An Enzyme’s Active Site Binds Substrates and Carries Out Catalysis
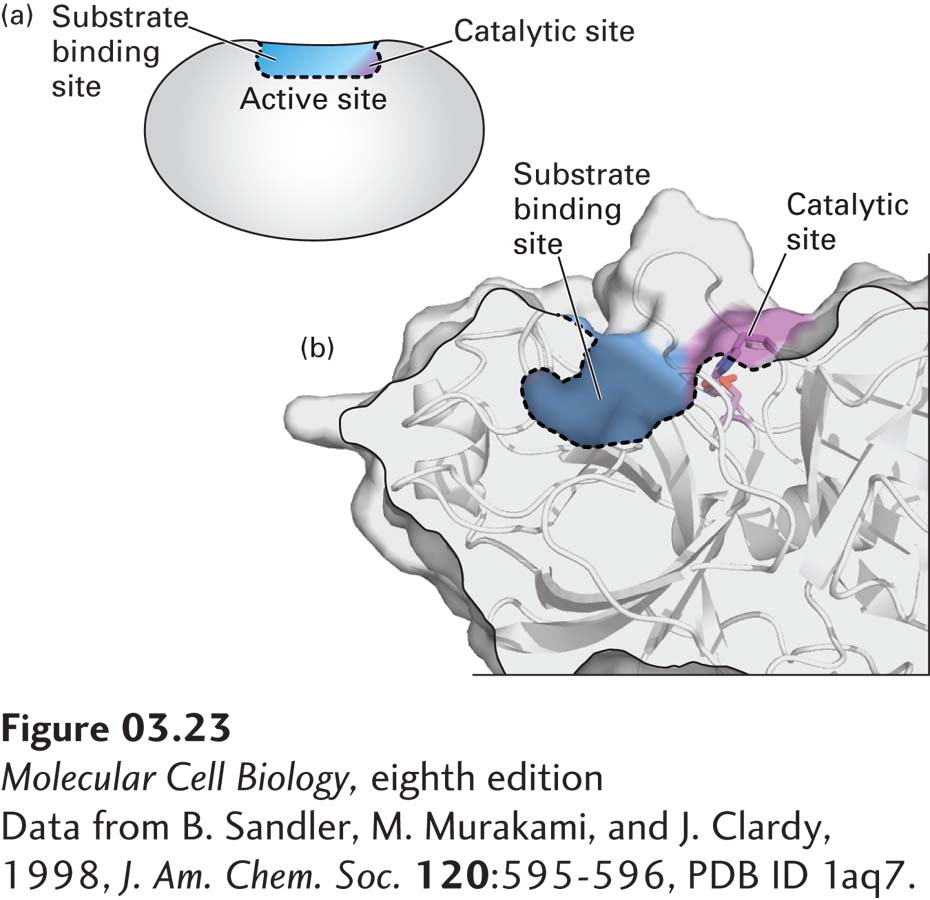
Certain amino acids of an enzyme are particularly important in determining its specificity and catalytic power. In the native conformation of an enzyme, critically important amino acids (which usually come from different parts of the linear sequence of the polypeptide) are brought into proximity, forming a cleft in the enzyme surface called the active site (Figure 3-23). An active site usually makes up only a small part of the total protein; the remaining part is involved in the folding of the polypeptide, regulation of the active site, and interactions with other molecules.
[Data from B. Sandler, M. Murakami, and J. Clardy, 1998, J. Am. Chem. Soc.120:595-596, PDB ID 1aq7.]
FIGURE 3-23Active site of the enzyme trypsin. (a) An enzyme’s active site (outlined by dashed line) is composed of a substrate-binding site (blue), which binds specifically to a substrate, and a catalytic site (purple), which carries out catalysis. (b) A hybrid surface/ribbon representation of a portion of the serine protease trypsin. Clearly visible are the active-site cleft containing the catalytic site (purple, includes the key catalytic triad of Ser-195, Asp-102, and His-57, see also Figure 3-27) and a portion of the substrate-binding site called the side-chain-specificity binding pocket (blue).
[Data from B. Sandler, M. Murakami, and J. Clardy, 1998, J. Am. Chem. Soc.120:595-596, PDB ID 1aq7.]
An active site consists of two functionally important regions: the substrate-binding site, which recognizes and binds the substrate or substrates, and the catalytic site, which carries out the chemical reaction once the substrate has bound. The catalytic groups in the catalytic site are amino acid side chains and backbone carboxyl and amino groups. In some enzymes, the catalytic and substrate-binding sites overlap; in others, the two regions are structurally distinct.
The substrate-binding site is responsible for the remarkable specificity of enzymes. Alteration of the structure of an enzyme’s substrate by only one or a few atoms, or a subtle change in the geometry (e.g., stereochemistry) of the substrate, can result in a variant molecule that is no longer a substrate of the enzyme. As with the specificity of antibodies for antigens described above, the specificity of enzymes for substrates is a consequence of the precise molecular complementarity between an enzyme’s substrate-binding site and the substrate. Usually only one or a few substrates can fit precisely into a binding site.
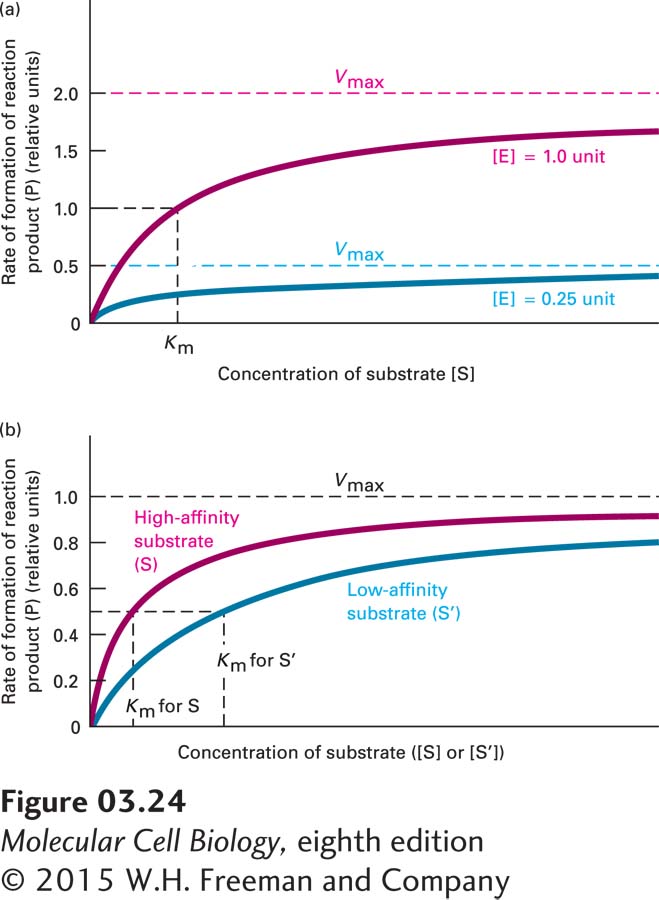
The idea that substrates might bind to enzymes in the manner of a key fitting into a lock was first suggested by Emil Fischer in 1894. A variation of this proposal by Daniel Koshland in 1958, called induced fit, posited that the substrate-binding site is not rigid, as a lock is, but flexible, and is induced to change shape for more optimal catalysis when the substrate binds. In 1913, Leonor Michaelis and Maud Leonora Menten provided crucial evidence supporting the enzyme-substrate binding hypothesis. They showed that the rate of an enzymatic reaction was proportional to the substrate concentration at low substrate concentrations, but that as substrate concentrations increased, the rate reached a plateau, or maximal velocity, Vmax, and became substrate concentration independent, with a value of Vmax directly proportional to the amount of enzyme present in the reaction mixture (Figure 3-24).
FIGURE 3-24Km and Vmax for an enzyme-catalyzed reaction.Km and Vmax are determined from analysis of the dependence of the initial reaction rate on substrate concentration. The shape of these hypothetical kinetic curves is characteristic of a simple enzyme-catalyzed reaction in which one substrate (S) is converted into product (P). The initial reaction velocity is measured immediately after addition of enzyme to substrate, before the substrate concentration changes appreciably. (a) Plots of initial reaction velocity at two different concentrations of enzyme [E] as a function of substrate concentration [S]. The [S] that yields a half-maximal reaction rate is the Michaelis constant Km, a measure of the affinity of E for turning S into P. Quadrupling the enzyme concentration causes a proportional increase in the reaction rate, so the maximal velocity Vmax is quadrupled; Km, however, is unaltered. (b) Plots of initial reaction velocity versus substrate concentration with a substrate S for which the enzyme has a high affinity and with a substrate S′ for which the enzyme has a lower affinity. Note that Vmax is the same with both substrates because [E] is the same, but that Km is higher for S′, the low-affinity substrate.
Page 92
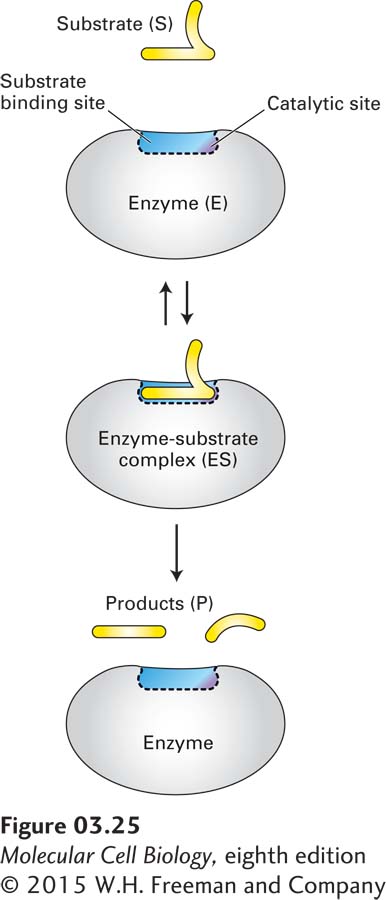
FIGURE 3-25Schematic model of an enzyme’s reaction mechanism. Enzyme kinetics suggest that enzymes (E) bind substrate molecules (S) at a fixed and limited number of sites –the enzymes’ active sites. The bound species is known as an enzyme-substrate (ES) complex. The ES complex is in equilibrium with the unbound enzyme and substrate (double arrows) and is an intermediate step in the conversion of substrate to products (P).
Michaelis and Menten deduced that these characteristics were due to the binding of substrate molecules (S) to a fixed and limited number of sites on the enzymes (E), and they called the bound species the enzyme-substrate (ES) complex. At high concentrations of substrate, all the binding sites on the enzymes have substrate bound, and the substrate-binding sites are said to be saturated with substrate—no additional binding to active sites is possible, and the maximal velocity of the reaction is achieved. Michaelis and Menten proposed that the ES complex is in equilibrium with the unbound enzyme and substrate and is an intermediate step in the ultimately irreversible conversion of substrate to product (P) (Figure 3-25):
E + S ⇌ ES → E + P
and that the rate V0 of formation of product at a particular substrate concentration [S] is given by what is now called the Michaelis-Menten equation:
where the Michaelis constant,Km, a measure of the affinity of an enzyme for its substrate, is the substrate concentration that yields a half-maximal reaction rate (i.e., ½ Vmax in Figure 3-24). The Km is somewhat similar in nature, but not identical, to the dissociation constant, Kd (see Chapter 2). The smaller the value of Km, the more effective the enzyme is at making product from dilute solutions of substrate, and the lower the substrate concentration needed to reach half-maximal velocity. The smaller the Kd, the lower the ligand concentration needed to reach 50 percent of binding. The concentrations of the various small molecules in a cell vary widely, as do the Km values for the different enzymes that act on them. A good rule of thumb is that the intracellular concentration of a substrate is often approximately the same as, or somewhat greater than, the Km value of the enzyme to which it binds.
The rates of reaction at substrate saturation vary enormously among enzymes. The maximum number of substrate molecules converted to product at a single enzyme active site per second, called the turnover number, can be less than 1 for very slow enzymes. The turnover number for carbonic anhydrase, one of the fastest enzymes, is 6 × 105 molecules per second.
Many enzymes catalyze the conversion of substrates to products by dividing the process into multiple, discrete chemical reactions, in which the product of one reaction is the substrate for the subsequent reaction. These sequential reactions generate multiple, distinct enzyme-substrate complexes (ES, ES′, ES″, etc.) prior to the final release of the products:
E + S ⇌ ES ⇌ ES′ ⇌ ES″ ⇌ …E + P
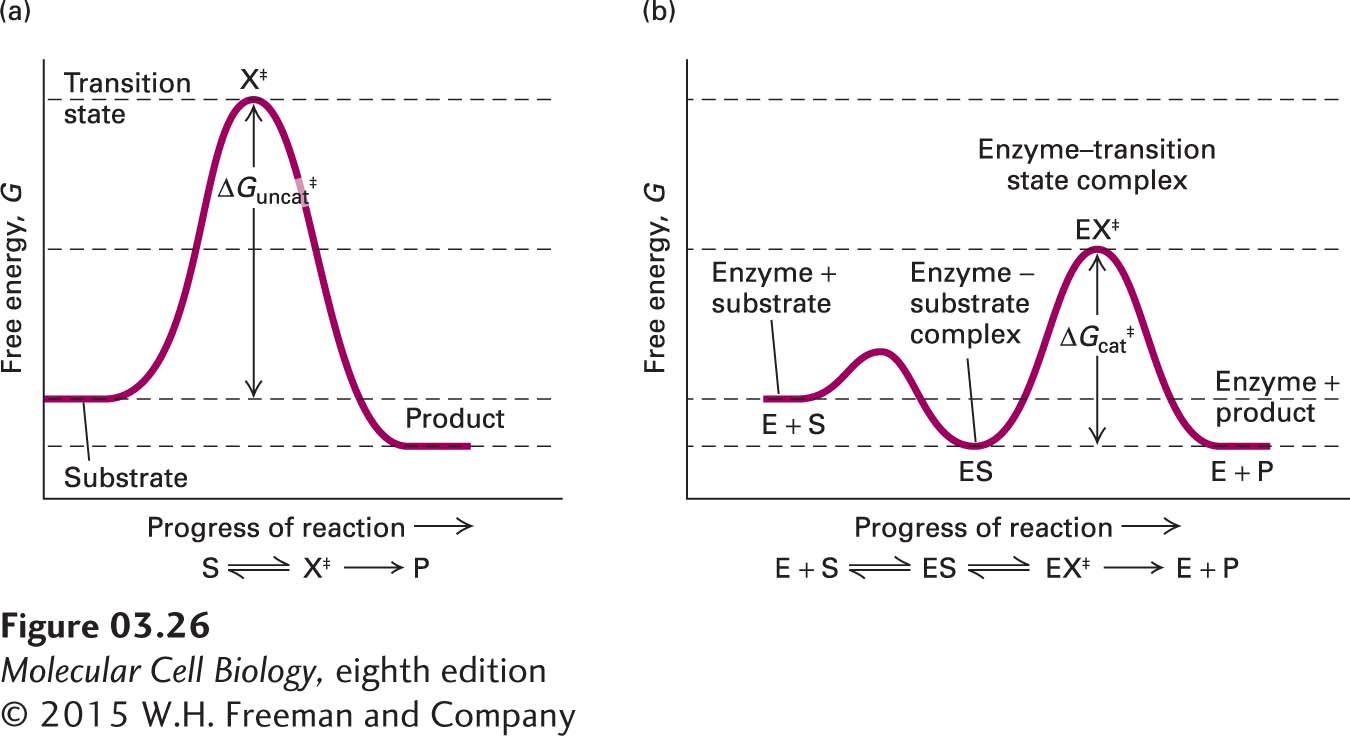
The energy profiles for such multistep reactions involve multiple hills and valleys (Figure 3-26). Methods have been developed to trap the intermediates in such reactions to learn more about the details of how enzymes catalyze reactions.
FIGURE 3-26Free-energy reaction profiles of uncatalyzed and multistep enzyme-catalyzed reactions. (a) The free-energy reaction profile of a hypothetical simple uncatalyzed reaction converting substrate (S) to product (P) via a single high-energy transition state. (b) Many enzymes catalyze such reactions by dividing the process into multiple discrete steps, in this case, the initial formation of an ES complex followed by conversion via a single transition state (EX‡) to the free enzyme (E) and P. The activation energy for each of these steps is significantly less than the activation energy for the uncatalyzed reaction; thus the enzyme dramatically enhances the reaction rate.