Electrophoresis Separates Molecules on the Basis of Their Charge-to-Mass Ratio
Electrophoresis, a technique for separating molecules in a mixture under the influence of an applied electric field, is one of the most frequently used techniques to study proteins and nucleic acids. Dissolved molecules in an electric field move, or migrate, at a speed determined by their charge-to-mass (charge:mass) ratio and the physical properties of the medium through which they migrate. For example, if two molecules have the same mass and shape, the one with the greater net charge will move faster toward an electrode of the opposite polarity.
SDS-Polyacrylamide Gel Electrophoresis Because many proteins or nucleic acids that differ in size and shape have nearly identical charge:mass ratios, electrophoresis of these macromolecules in a liquid solution results in little or no separation of molecules of different lengths. However, successful separation of proteins and nucleic acids can be accomplished by electrophoresis in various gels (semisolid suspensions in water similar to the congealed gelatin found in desserts). These gels are commonly cast into flat, relatively thin slabs between a pair of glass plates. When a mixture of proteins is placed in a gel and an electric current is applied, the gel acts as a sieve, allowing smaller species to maneuver more rapidly through its pores than larger species do. The shape of a molecule can also influence its rate of migration (long asymmetric molecules migrate more slowly than spherical ones of the same mass).
Electrophoretic separation of proteins is most commonly performed in polyacrylamide gels. These gels are made by polymerizing a solution of acrylamide monomers into polyacrylamide chains and simultaneously cross-linking the chains into a semisolid matrix. The pore size of a gel can be varied by adjusting the concentrations of polyacrylamide and the cross-linking reagent. The rate at which a protein moves through a gel is influenced by the gel’s pore size and the strength of the electric field. By suitable adjustment of these parameters, proteins of widely varying sizes can be resolved (separated from one another) by this technique, known as polyacrylamide gel electrophoresis (PAGE).
Page 108
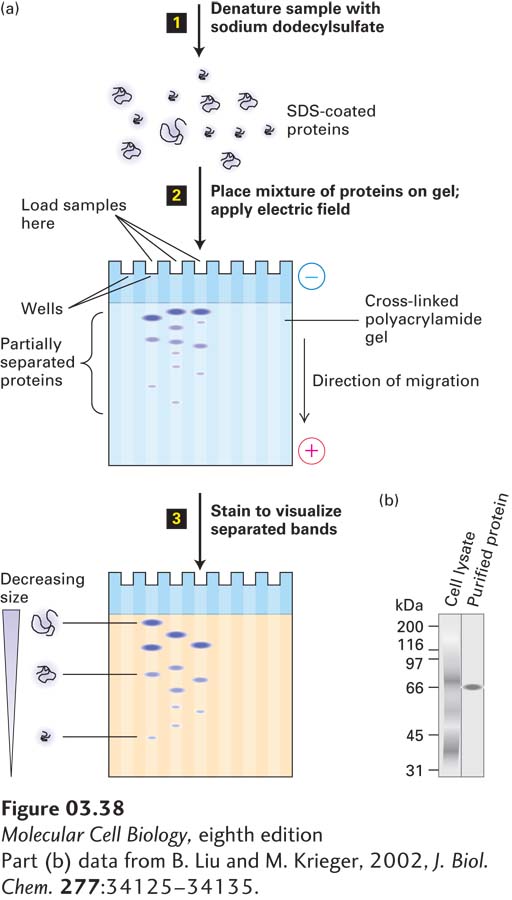
In the most powerful technique for resolving protein mixtures, proteins are exposed to the ionic detergent SDS (sodium dodecylsulfate) before and during gel electrophoresis (Figure 3-38). SDS denatures proteins, in part because it binds to hydrophobic side chains, destabilizing the hydrophobic interactions in the core of a protein that contribute to its stable conformation. SDS treatment is usually combined with heating, often in the presence of reducing agents that break disulfide bonds. Under these conditions, most multimeric proteins dissociate into their subunits. Typically, the amount of SDS that binds to the protein is proportional to the length of the polypeptide chain and relatively independent of the sequence. Two proteins of similar size will bind the same absolute quantity of SDS, whereas a protein twice that size will bind twice the amount of SDS. Denaturation of a complex protein mixture with SDS in combination with heat usually forces each polypeptide chain into an extended conformation and imparts on each of the proteins in the mixture a constant charge:mass ratio because the dodecylsulfate, which is negatively charged, is the major contributor of charge. As the SDS-bound proteins move through the polyacrylamide gel, they are separated according to size by the sieving action of the gel. SDS treatment thus eliminates the effect of differences in native conformation; therefore, chain length, which is proportional to mass, is the principal determinant of the migration rate of proteins in SDS-polyacrylamide electrophoresis (SDS-PAGE). Even chains that differ in molecular weight by less than 10 percent can be resolved by this technique. Moreover, the molecular weight of a protein can be estimated by comparing the distance that it migrates through a gel with the distances that proteins of known molecular weight (called molecular weight “standards”) migrate in the same gel (there is a roughly linear relationship between migration distance and the log of the molecular weight). Proteins within the gels can be extracted for further analysis (e.g., identification by the methods described below).
[Part (b) data from B. Liu and M. Krieger, 2002, J. Biol. Chem. 277:34125–34135.]
EXPERIMENTAL FIGURE 3-38SDS-polyacrylamide gel electrophoresis (SDS-PAGE) separates proteins primarily on the basis of their masses. (a) Initial treatment with SDS, a negatively charged detergent, dissociates multimeric proteins and denatures all the polypeptide chains (step 1). During electrophoresis, the SDS-protein complexes migrate through the polyacrylamide gel (step 2). Small complexes are able to move through the pores faster than larger ones. Thus the proteins separate into bands according to their sizes as they migrate. The separated protein bands are visualized by staining with a dye (step 3). (b) Example of SDS-PAGE separation of all the proteins in a whole-cell lysate (detergent-solubilized cells). (Left) The many separate stained proteins appear almost as a continuum. (Right) A single protein purified from the lysate by a single step of antibody-affinity chromatography. The proteins were visualized by staining with a silver-based dye.
[Part (b) data from B. Liu and M. Krieger, 2002, J. Biol. Chem. 277:34125–34135.]
If two or more polypeptides are cross-linked by disulfide bonds, the protein’s migration rate in SDS-PAGE will depend on whether or not the protein has been reduced to break those bonds prior to electrophoresis. The cross-linked proteins will appear larger than the individual, reduced subunits. By examining samples with and without reduction, one can identify such proteins and their component polypeptides.
Two-Dimensional Gel Electrophoresis Electrophoresis of a mixture containing all cellular proteins by SDS-PAGE can separate proteins having relatively large differences in mass, but cannot readily resolve proteins having similar masses (e.g., a 41-kDa protein versus a 42-kDa protein). To separate proteins of similar masses, another physical characteristic must be exploited. Most commonly, this characteristic is electric charge, which is determined by the pH of the sample and by the relative number of the protein’s positively and negatively charged groups, which is in turn dependent on the pKa’s of the ionizable groups (see Chapter 2) on the proteins (usually the amino and carboxyl termini and side chains such as those in lysine and aspartic acid). Two unrelated proteins having similar masses are unlikely to have identical net charges because their sequences, and thus the number of acidic and basic residues, are different.
Page 109
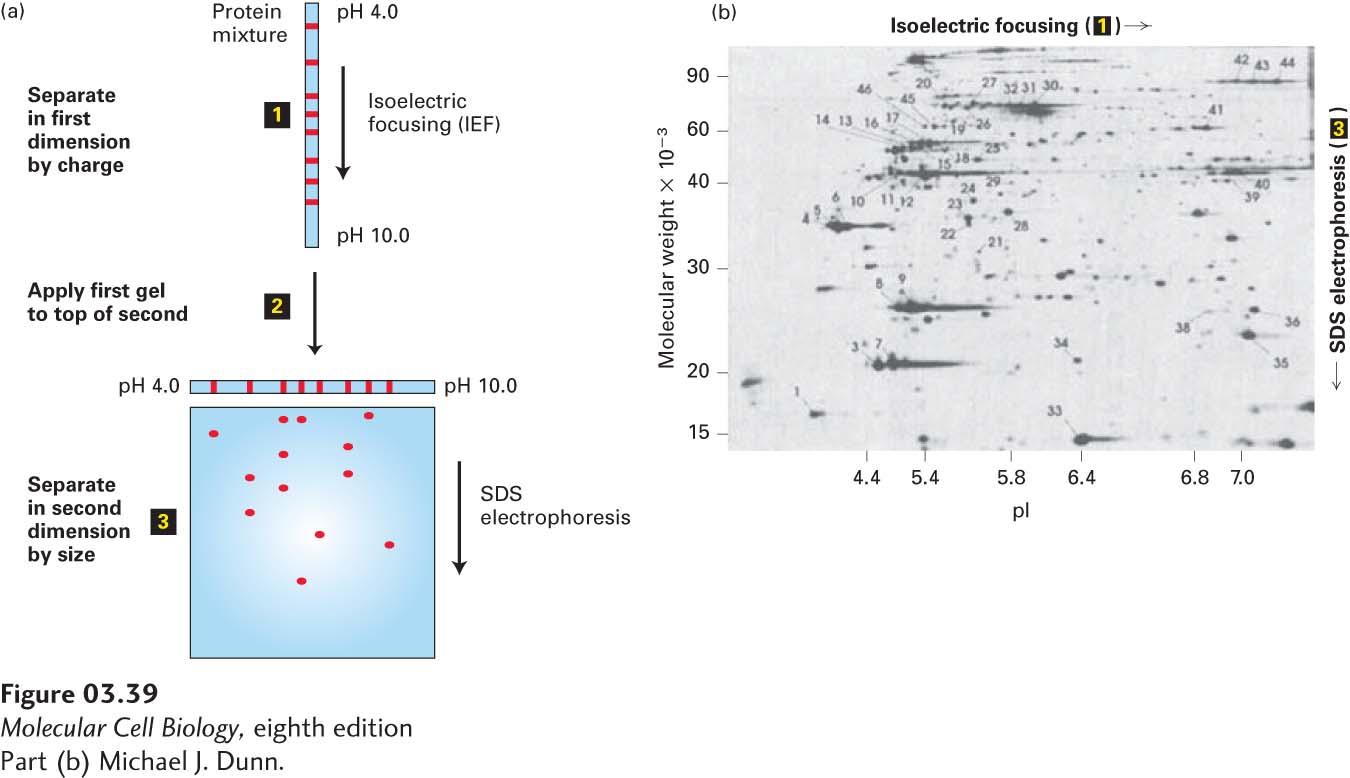
In two-dimensional gel electrophoresis, proteins are separated sequentially, first by their charges and then by their masses (Figure 3-39a). In the first step, a cell or tissue extract is fully denatured by high concentrations (8 M) of urea (and sometimes SDS) and then layered on a strip of gel that contains urea, which removes any bound SDS, and a continuous pH gradient. The pH gradient is formed by ampholytes, polyanionic and polycationic small molecules that are cast into the gel. When an electric field is applied to the gel, the ampholytes will migrate. Ampholytes with an excess of negative charges will migrate toward the anode, where they establish an acidic pH (many protons), while ampholytes with an excess of positive charges will migrate toward the cathode, where they establish an alkaline pH. The careful choice of the mixture of ampholytes and careful preparation of the gel allows the construction of stable pH gradients ranging from pH 3 to pH 10. A charged protein placed at one end of such a gel will migrate through the gradient under the influence of the electric field until it reaches its isoelectric point (pI), the pH at which the net charge of the protein is zero. With no net charge, the protein will migrate no further. This technique, called isoelectric focusing (IEF), can resolve proteins that differ by only one charge unit. This method is sensitive enough to separate phosphorylated and nonphosphorylated versions of the same protein.
[Part (b) Michael J. Dunn.]
EXPERIMENTAL FIGURE 3-39Two-dimensional gel electrophoresis separates proteins on the basis of charge and mass. (a) In this technique, proteins are first separated into bands on the basis of their charges by isoelectric focusing (step 1). The resulting gel strip is applied to an SDS-polyacrylamide gel (step 2), and the proteins are separated into spots by mass (step 3). (b) In this two-dimensional electrophoresis gel of a protein extract from cultured cells, each spot represents a single polypeptide. Polypeptides can be detected by dyes, as here, or by other techniques, such as autoradiography. Each polypeptide is characterized by its isoelectric point (pI) and molecular weight.
[Part (b) Michael J. Dunn.]
Proteins that have been separated on an IEF gel can then be separated in a second dimension on the basis of their molecular weights. To accomplish this separation, the IEF gel strip is placed lengthwise on one outside edge of a square or rectangular slab of polyacrylamide gel, this time saturated with SDS to confer on each separated protein a more or less constant charge:mass ratio. When an electric field is imposed, the proteins will migrate from the IEF gel into the SDS gel and then separate according to their masses. The sequential resolution of proteins by charge and mass can achieve excellent separation of cellular proteins and provides a powerful visual representation of the complexity of proteins in cells (Figure 3-39b). Today sophisticated mass spectrometry methods, described below, are often used in place of two-dimensional gel electrophoresis, both to separate and to identify the protein components of a complex sample as well as to compare changes in the amounts of those components in different biological specimens.