Immunofluorescence Microscopy Can Detect Specific Proteins in Fixed Cells
The common chemical dyes mentioned above stain nucleic acids or broad classes of proteins, but it is much more informative to detect the presence and location of specific proteins. Immunofluorescence microscopy, the most widely used method of detecting specific proteins, uses an antibody to which a fluorescent dye has been covalently attached. To use this method, one must first generate antibodies to the specific protein of interest. As discussed briefly in Section 4.1 and in detail in Chapter 23, as part of the response to infection, the vertebrate immune system generates proteins called antibodies that bind specifically to the infectious agent. Cell biologists have made use of this immunological response to generate antibodies to specific proteins. Consider you have purified protein X and then inject it into an experimental animal so that it responds to the protein as a foreign molecule. Over a period of weeks, the animal will mount an immune response and make antibodies to protein X (the “antigen”). If you collect the blood from the animal, it will have antibodies to protein X mixed in with antibodies to many other different antigens, together with all the other blood proteins. You can now covalently bind protein X to a resin and, using affinity chromatography, bind and selectively retain just those antibodies specific to protein X. The antibodies can be eluted from the resin, and now you have a reagent that binds specifically to protein X. This approach generates polyclonal antibodies since many different cells in the animal have contributed the antibodies, and the antibodies are likely to bind to many different epitopes on protein X. Alternatively, as we described earlier in this chapter, it is possible to generate a clonal cell line that secretes antibodies to a specific epitope on protein X; these are called monoclonal antibodies.
To use either type of antibody to localize a protein, the cells or tissue must first be fixed to ensure that all components remain in place, and the cell must be permeabilized to allow entry of the antibody, which is commonly done by incubating the cells with a non-ionic detergent or by extracting the lipids with an organic solvent. In one version of immunofluorescence microscopy, the antibody is covalently linked to a fluorochrome. Classically used fluorochromes include rhodamine and Texas red, which emit red light; Cy3, which emits orange light; and fluorescein, which emits green light; but newer and more photostable fluorochromes, with emission wavelengths from blue to far-red, have now been developed. When a fluorochrome-antibody complex is added to a permeabilized cell or tissue section, the complex will bind to the corresponding antigen, then light up when illuminated at the excitation wavelength. Staining a specimen with different dyes that fluoresce at different wavelengths allows multiple proteins as well as DNA to be localized within the same cell (see the chapter-opening figure).
Page 145
The most commonly used variation of this technique is called indirect immunofluorescence microscopy because the antibody specific to the protein of interest is detected indirectly. In this technique, an unlabeled monoclonal or polyclonal antibody is applied to the specimen, followed by a second, fluorochrome-tagged antibody that binds to the constant (Fc) segment of the first antibody. For example, a “secondary” antibody can be generated by immunizing a goat with the Fc segment that is common to all rabbit IgG antibodies; when coupled to a fluorochrome, this second antibody preparation (called “goat anti-rabbit”) will detect any rabbit antibody used to stain a tissue or cell (Figure 4-14). Because several goat anti-rabbit antibody molecules can bind to a single rabbit antibody molecule in a specimen, the fluorescence is generally much brighter than if a single fluorochrome-tagged antibody were used. This approach is often extended to do double-label fluorescence microscopy, in which two proteins can be visualized simultaneously. For example, two proteins can be visualized by indirect immunofluorescence microscopy using primary antibodies made in different animals (e.g., rabbit and chicken) and secondary antibodies (e.g., goat–anti-rabbit and sheep–anti-chicken) labeled with different fluorochromes. In another variation, one protein can be visualized by indirect immunofluorescence microscopy and the second protein by a dye that specifically binds to it. Once the individual images are taken on the fluorescence microscope, they can be merged electronically (Figure 4-15).
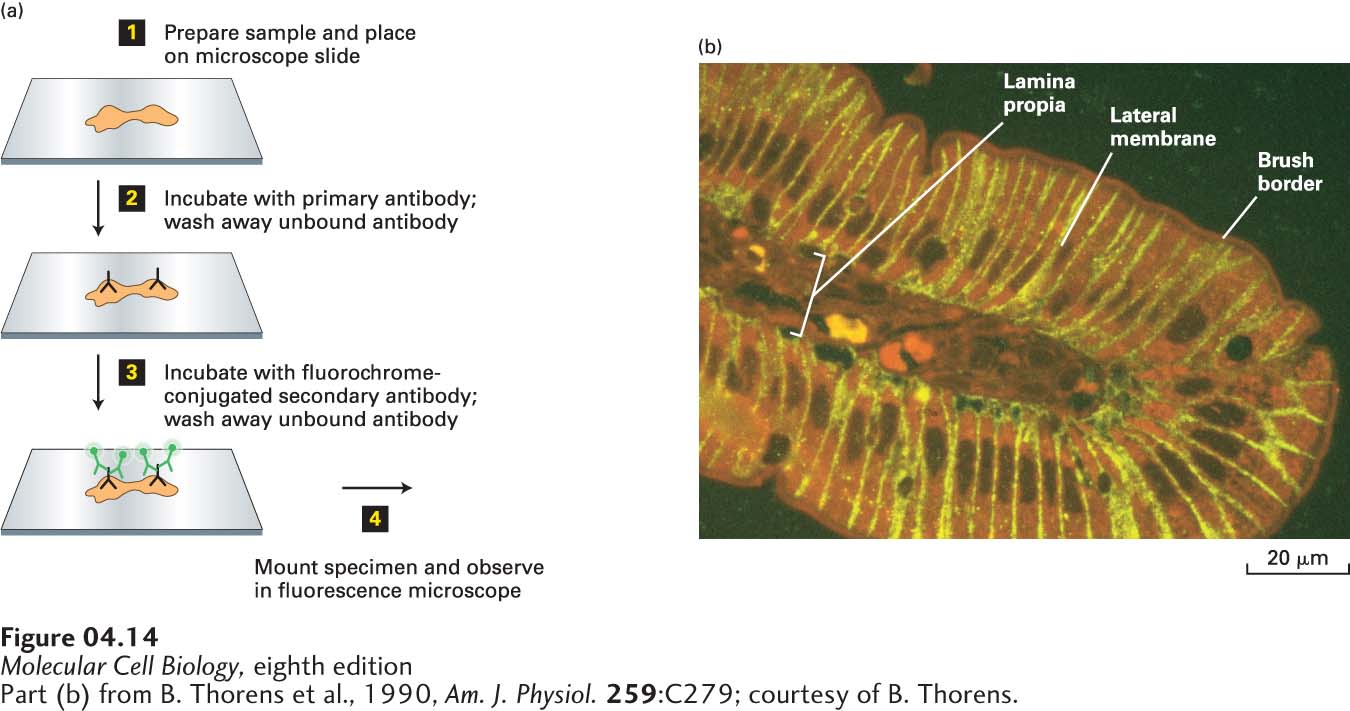
[Part (b) from B. Thorens et al., 1990, Am. J. Physiol.259:C279; courtesy of B. Thorens.]
FIGURE 4-14A specific protein can be localized in fixed tissue sections by indirect immunofluorescence microscopy. (a) To localize a protein by immunofluorescence microscopy, a tissue section, or sample of cells, must be chemically fixed and made permeable to antibodies (step 1). The sample is then incubated with a primary antibody that binds specifically to the antigen of interest, and unbound antibody is then removed by washing (step 2). The sample is next incubated with a fluorochrome-labeled secondary antibody that specifically binds to the primary antibody, and again, excess secondary antibody is removed by washing (step 3). The sample is then mounted in specialized mounting medium and examined in a fluorescence microscope (step 4). (b) In this example, a section of the rat intestinal wall was stained with Evans blue, which generates a nonspecific red fluorescence, and GLUT2, a glucose transport protein, was localized by indirect immunofluorescence microscopy. GLUT2 (yellow) is seen to be present in the basal and lateral sides of the intestinal cells, but is absent from the brush border, composed of closely packed microvilli on the apical surface facing the intestinal lumen. Capillaries run through the lamina propria, a loose connective tissue beneath the epithelial layer.
[Part (b) from B. Thorens et al., 1990, Am. J. Physiol.259:C279; courtesy of B. Thorens.]
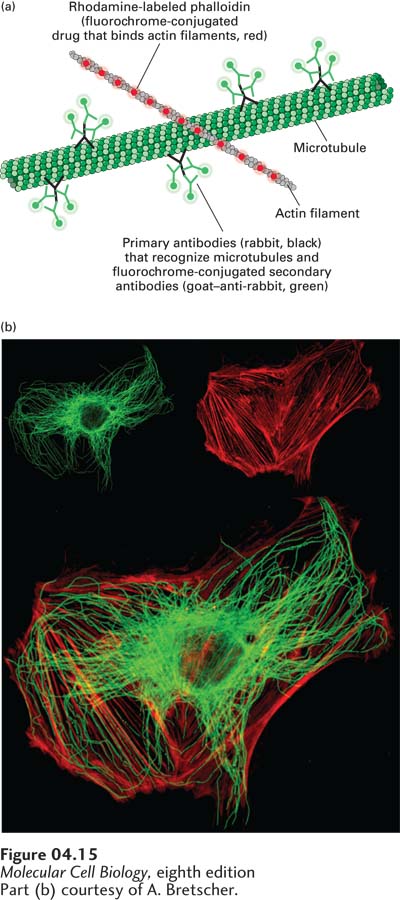
[Part (b) courtesy of A. Bretscher.]
EXPERIMENTAL FIGURE 4-15Double-label fluorescence microscopy can visualize the relative distributions of two proteins. In double-label fluorescence microscopy, each protein must be labeled with a different fluorochrome. (a) A cultured cell was fixed and permeabilized and then incubated with Rhodamine-labeled phalloidin, a reagent that specifically binds to filamentous actin. It was also incubated with rabbit antibodies to tubulin, the major component of microtubules, followed by a fluorescein-labeled secondary goat–anti-rabbit antibody. (b) The upper panels show the fluorescein-stained tubulin (left) and Rhodamine-stained actin (right), and the lower panel shows the electronically merged images.
[Part (b) courtesy of A. Bretscher.]
In another widely used version of this technology, molecular genetic techniques are used to make a cDNA encoding a recombinant protein to which is fused a short sequence of amino acids called an epitope tag. When expressed in cells, this cDNA will generate the protein linked to the specific tag. Two commonly used epitope tags are FLAG, which encodes the amino acid sequence DYKDDDDK (single-letter amino acid code), and myc, which encodes the sequence EQKLISEEDL. Commercial fluorochrome-coupled monoclonal antibodies to the FLAG or myc epitopes can then be used to detect the recombinant protein in the cell. In an extension of this technology to allow the simultaneous visualization of two proteins, one protein can be tagged with FLAG and a different protein with myc. Each tagged protein is then visualized with a different color, for example, with an Alexa-488-labeled antibody (emitting green light) to the myc epitope and an Alexa-568-labeled antibody (emitting red light) to the FLAG epitope.