The Polymerase Chain Reaction Amplifies a Specific DNA Sequence from a Complex Mixture
If the nucleotide sequences at the ends of a particular DNA region are known, the intervening fragment can be amplified directly by the polymerase chain reaction (PCR). Here we describe the basic PCR technique and three situations in which it is used.
Page 240
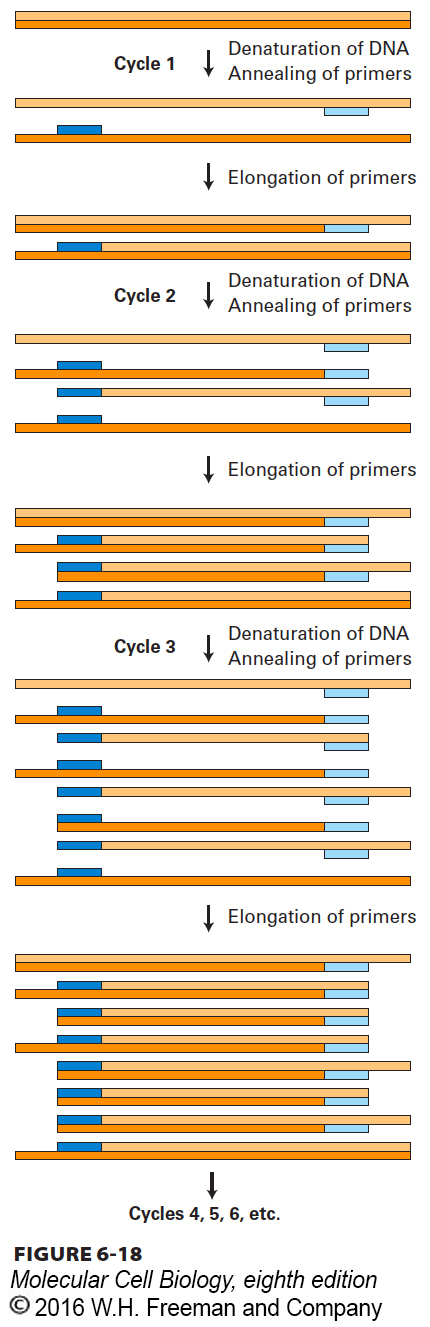
The PCR depends on the ability to alternately denature (melt) double-stranded DNA molecules and hybridize complementary single strands in a controlled fashion. As outlined in Figure 6-18, a typical PCR procedure begins with heat-denaturation of a DNA sample into single strands at 95 °C. Next, two synthetic oligonucleotides complementary to the 3′ ends of the DNA segment of interest (the target sequence) are added in great excess to the denatured DNA, and the temperature is lowered to 50–60 °C. These specific oligonucleotides, which are present at a very high concentration, hybridize to their complementary sequences in the DNA sample, whereas the long strands of the sample DNA remain apart because of their comparatively low concentration. The hybridized oligonucleotides then serve as primers for DNA chain synthesis in the presence of deoxynucleotides (dNTPs) and a temperature-resistant DNA polymerase such as that from Thermus aquaticus, a bacterium that lives in hot springs. This enzyme, called Taq polymerase, can remain active even after being heated to 95 °C and can extend the primers at temperatures up to 72 °C. When synthesis is complete, the whole mixture is reheated to 95 °C to denature the newly formed DNA duplexes. After the temperature is lowered again, another cycle of synthesis takes place because excess primer is still present. Repeated cycles of denaturation (heating) followed by hybridization and synthesis (cooling) quickly amplify the sequence of interest. At each cycle, the number of copies of the sequence between the primer sites is doubled; therefore, the target sequence increases exponentially—about 1-million-fold after 20 cycles—whereas all other sequences in the original DNA sample remain unamplified.
FIGURE 6-18The polymerase chain reaction (PCR) is widely used to amplify DNA regions with known flanking sequences. To amplify a specific region of DNA, an investigator chemically synthesizes two different oligonucleotide primers complementary to sequences of approximately 18 bases flanking the region of interest (shown here as light blue and dark blue bars). The complete reaction is composed of a complex mixture of double-stranded DNA (usually genomic DNA containing the target sequence of interest), a stoichiometric excess of both primers, the four dNTPs, and a heat-stable DNA polymerase known as Taq polymerase. During each PCR cycle, the reaction mixture is first heated to separate the strands and then cooled to allow the primers to bind to complementary sequences flanking the region to be amplified. Taq polymerase then extends each primer from its 3′ end, generating newly synthesized strands that extend in the 3′ direction to the 5′ end of the template strand. During the third cycle, two double-stranded DNA molecules are generated equal in length to the sequence of the region to be amplified. In each successive cycle, the target sequence, which anneals to the primers, is duplicated, so it eventually vastly outnumbers all other DNA sequences in the reaction mixture. Successive PCR cycles can be automated by cycling the reaction at timed intervals between a high temperature for DNA melting and a lower temperature for the annealing and elongation parts of the cycle. A reaction that cycles 20 times will amplify the specific target sequence 1-million-fold.
Page 241
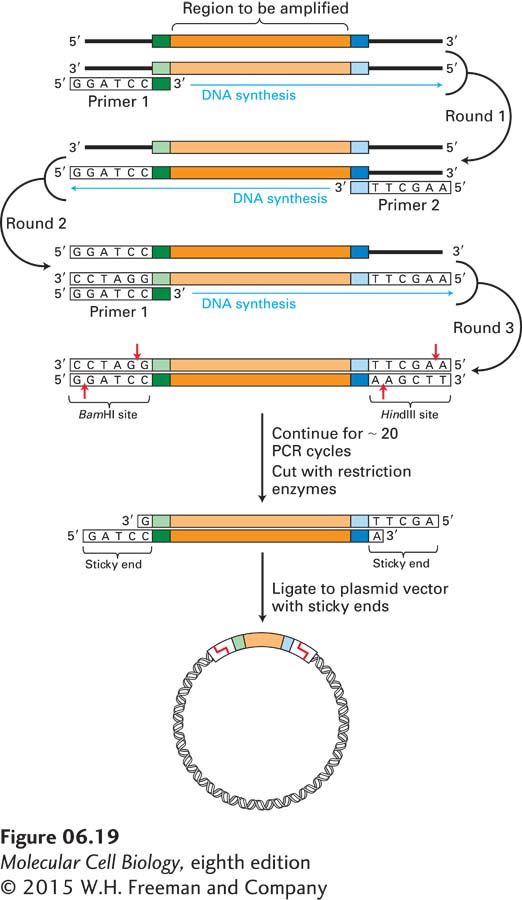
Direct Isolation of a Specific Segment of Genomic DNA For organisms in which all or most of the genome has been sequenced, PCR amplification starting with the total genomic DNA is often the easiest way to obtain a specific DNA region of interest for cloning. In this application of the PCR, the two oligonucleotide primers are designed to hybridize to sequences flanking the genomic region of interest and to include sequences that are recognized by specific restriction enzymes (Figure 6-19). For an oligonucleotide to be useful as a PCR primer, it must be long enough for its sequence to occur uniquely in the genome. For most purposes, this condition is satisfied by oligonucleotides containing about 20 nucleotides. Any given 20-nucleotide sequence occurs once in every 420 (∼1012) nucleotides by chance, so it is usually possible to identify two specific 20-nucleotide sequences flanking the target sequence that each occur only once in the genome.
EXPERIMENTAL FIGURE 6-19A specific target sequence in genomic DNA can be amplified by PCR for use in cloning. Each primer for PCR is complementary to one end of the target sequence and includes the recognition site for a restriction enzyme that does not have a recognition site within the target region. In this example, primer 1 contains a BamHI recognition sequence, whereas primer 2 contains a HindIII recognition sequence. (Note that for clarity, in any round, amplification of only one of the two strands—the one in brackets—is shown.) After amplification, the target segments are treated with appropriate restriction enzymes, generating fragments with sticky ends. These fragments can be incorporated into complementary plasmid vectors and cloned in E. coli by the usual procedure (see Figure 6-14).
After amplification of the target sequence for about 20 PCR cycles, cleavage with the appropriate restriction enzymes produces sticky ends that allow efficient ligation of the fragment to a plasmid vector whose polylinker has been cleaved by the same restriction enzymes. The resulting recombinant plasmids, all carrying the identical genomic DNA segment, can then be cloned in E. coli cells. With certain refinements of the PCR, even DNA segments greater than 10 kb in length can be amplified and cloned in this way.
Page 242
Note that this method does not require the cloning of large numbers of restriction fragments derived from genomic DNA and their subsequent screening to identify the specific fragment of interest. In effect, the PCR method inverts this traditional approach and so avoids its most tedious aspects. The PCR method is useful for isolating gene sequences to be manipulated in a variety of useful ways described later. In addition, the PCR method can be used to isolate gene sequences from mutant organisms to determine how they differ from the wild type.
A variation on the PCR method allows PCR amplification of a specific cDNA sequence made from cellular mRNAs. This method, known as reverse transcriptase–PCR (RT-PCR), begins with the same procedure described previously for isolation of cDNA from a collection of cellular mRNAs. Typically, an oligo-dT primer, which hybridizes to the 3′ poly(A) tail of the mRNA, is used as the primer for the first strand of cDNA synthesis by reverse transcriptase. A specific cDNA can then be isolated from this complex mixture of cDNAs by PCR amplification using two oligonucleotide primers designed to match sequences at the 5′ and 3′ ends of the corresponding mRNA. As described previously, these primers can be designed to include restriction sites to facilitate the insertion of the amplified cDNA into a suitable plasmid vector.
RT-PCR can be performed so that the starting amount of a particular cellular mRNA can be determined accurately. To carry out quantitative RT-PCR, the amount of double-stranded DNA sequence produced by each amplification cycle is determined as the amplification of a particular mRNA sequence proceeds. By extrapolation from these amounts, an estimate of the starting amount of the mRNA sequence can be obtained. Such quantitative RT-PCR analyses carried out on tissues or whole organisms using primers targeted to genes of interest provide one of the most accurate means to follow changes in gene expression.
Tagging of Genes by Insertion Mutations Another useful application of the PCR is the amplification of a “tagged” gene from the genomic DNA of a mutant strain. This approach is a simpler method for identifying genes associated with a particular mutant phenotype than the screening of a library by functional complementation (see Figure 6-16).
The key to this use of the PCR is the ability to produce mutations by inserting a known DNA sequence into the genome of an experimental organism. Such insertion mutations can be generated by the use of DNA transposons, which can move (or transpose) from one chromosomal site to another. As discussed in more detail in Chapter 8, these DNA sequences occur naturally in the genomes of most organisms and may give rise to loss-of-function mutations if they transpose into a protein-coding region.
Page 243
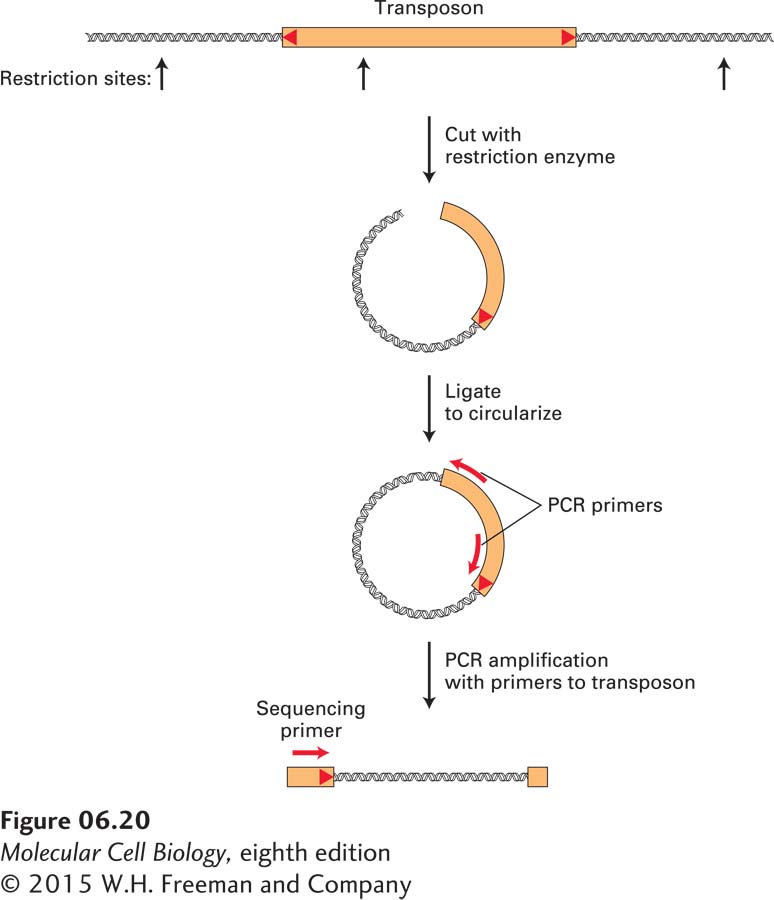
For example, researchers have modified a Drosophila DNA transposon, known as the P element, to optimize its use in the experimental generation of insertion mutations. Once it has been demonstrated that insertion of a P element causes a mutation with an interesting phenotype, the genomic sequences adjacent to the insertion site can be amplified by a variation of the standard PCR protocol that uses synthetic primers complementary to the known P-element sequence, but allows unknown neighboring sequences to be amplified. One such method, depicted in Figure 6-20, begins by cleaving Drosophila genomic DNA containing a P-element insertion with a restriction enzyme that makes a single cut within the P-element DNA. The resulting collection of cleaved DNA fragments, when treated with DNA ligase, yields circular molecules, some of which contain P-element DNA. The chromosomal region flanking the P element can then be amplified by PCR using primers that match P-element sequences and are elongated in opposite directions. The sequence of the resulting amplified fragment can then be determined using a third DNA primer. The crucial sequence for identifying the site of P-element insertion is the junction between the end of the P-element and genomic sequences. Overall, this approach avoids the cloning of large numbers of DNA fragments and their screening to detect a cloned DNA corresponding to a mutated gene of interest.
FIGURE 6-20The genomic sequence at the insertion site of a DNA transposon is revealed by PCR amplification and sequencing. To obtain the DNA sequence of the insertion site of a P element, it is necessary to amplify the junction between known transposon sequences and unknown flanking chromosomal sequences. One way to achieve this is to cleave genomic DNA with a restriction enzyme that cuts once within the transposon sequence. Ligation of the resulting restriction fragments will generate circular DNA molecules. By using appropriately designed DNA primers that match transposon sequences, it is possible to amplify the desired junction fragment using PCR. Finally, a DNA sequencing reaction (see Figures 6-21 and 6-22) is performed using the amplified fragment as a template and an oligonucleotide primer that matches sequences near the end of the transposon to obtain the sequence of the junction between the transposon and chromosome.
Similar methods have been applied to other organisms for which insertion mutations can be generated using either DNA transposons or viruses with sequenced genomes that can insert randomly into the genome.