Modifications of Histone Tails Control Chromatin Condensation and Function
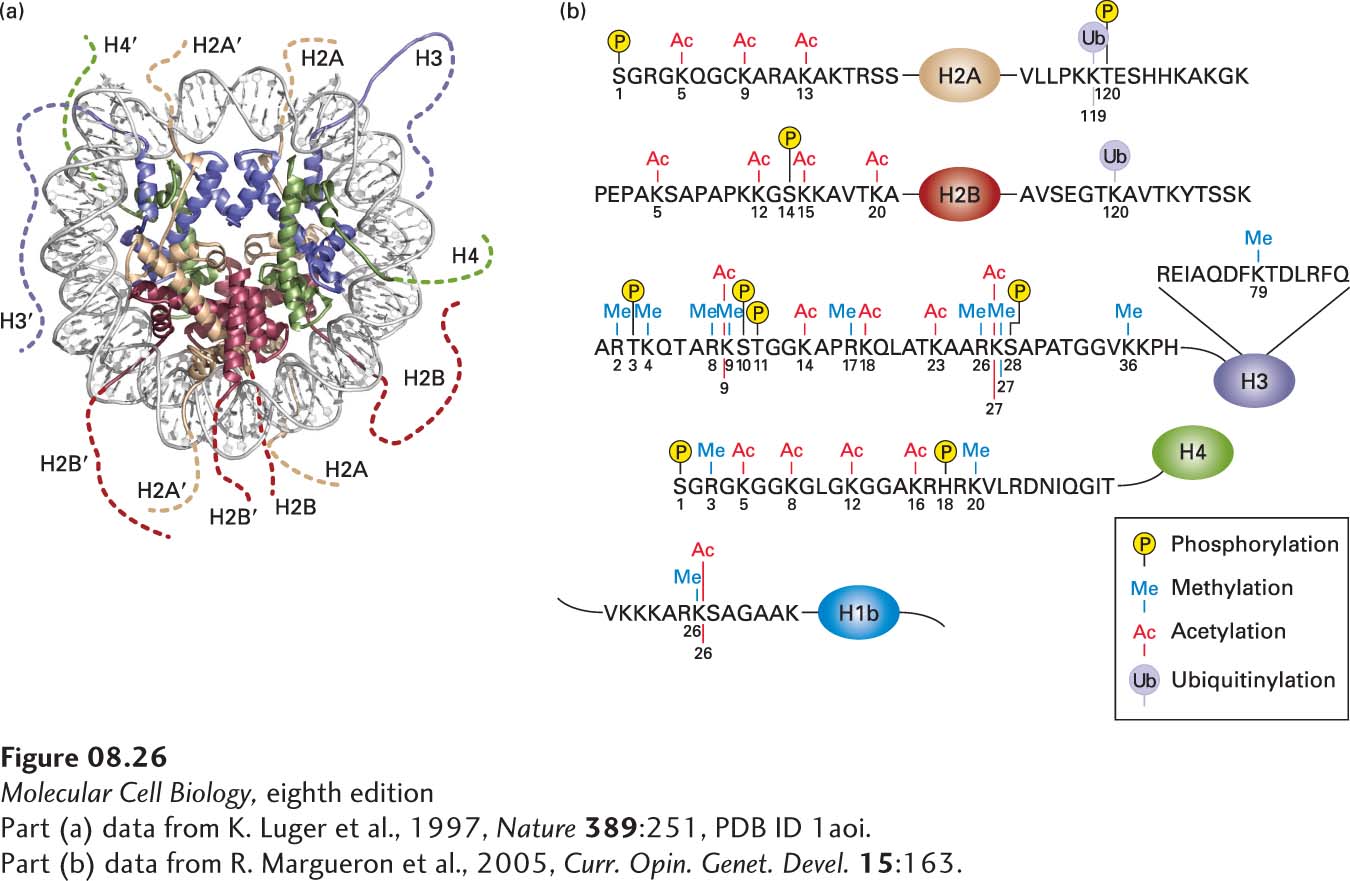
Each of the histone proteins making up the nucleosome core contains an intrinsically disordered, flexible N-terminus (see Figure 3-8) of 19–39 residues extending from the globular structure of the nucleosome; the H2A and H2B proteins also contain a flexible C-terminus extending from the globular histone octamer core. These termini, called histone tails, are represented in the model shown in Figure 8-26a. The histone tails are required for chromatin to condense from the beads-on-a-string conformation into the 30-nm fiber. For example, recent experiments indicate that the N-terminal tails of histone H4, particularly lysine16, are critical for forming the 30-nm fiber. This positively charged lysine interacts with a negative patch at the H2A–H2B interface of the next nucleosome in the stacked nucleosomes of the 30-nm fiber.
[Part (a) data from K. Luger et al., 1997, Nature389:251, PDB ID 1aoi. Part (b) data from R. Margueron et al., 2005, Curr. Opin. Genet. Devel.15:163.]
FIGURE 8-26Post-translational modifications observed on human histones. (a) Model of a nucleosome viewed from the top with histones shown as ribbon diagrams. This model depicts the lengths of the histone tails (dotted lines), which are not visible in the crystal structure (see Figure 8-24). The H2A N-terminal tails are at the bottom, and the H2A C-terminal tails are at the top. The H2B N-terminal tails are on the right and left, and the H2B C-terminal tails are at the bottom center. Histones H3 and H4 have short C-terminal tails that are not modified. (b) Summary of post-translational modifications observed in human histones. Histone-tail sequences are shown in the one-letter amino acid code (see Figure 2-14). The main portion of each histone is depicted as an oval. These modifications do not all occur simultaneously on a single histone molecule. Rather, specific combinations of a few of these modifications are observed on any one histone. See K. Luger and T. J. Richmond, 1998, Curr. Opin. Genet. Devel.8:140.
[Part (a) data from K. Luger et al., 1997, Nature389:251, PDB ID 1aoi. Part (b) data from R. Margueron et al., 2005, Curr. Opin. Genet. Devel.15:163.]
Histone tails are subject to multiple post-translational modifications such as acetylation, methylation, phosphorylation, and ubiquitinylation. Figure 8-26b summarizes the types of post-translational modifications observed in human histones. A particular histone protein never has all of these modifications simultaneously, but the histones in a single nucleosome usually contain several of these modifications simultaneously. The particular combinations of post-transcriptional modifications found in different regions of chromatin constitute a histone code that influences chromatin function by creating or removing binding sites for chromatin-associated proteins depending on the specific combinations of these modifications present. Here we describe the most abundant kinds of modifications found in histone tails and how these modifications control chromatin condensation and function. We end with a discussion of a special case of chromatin condensation, the inactivation of X chromosomes in female mammals.
Page 331
Histone Acetylation Histone-tail lysines undergo reversible acetylation and deacetylation by enzymes that act on specific lysines in the histone N-termini. In the acetylated form, the positive charge of the lysine ε-amino group is neutralized. As mentioned above, lysine 16 in histone H4 is particularly important for the folding of the 30-nm fiber because it interacts with a negatively charged patch on the surface of the neighboring nucleosome in the fiber. Consequently, when H4 lysine 16 is acetylated, the chromatin tends to form the less condensed “beads-on-a-string” conformation conducive for transcription and replication.
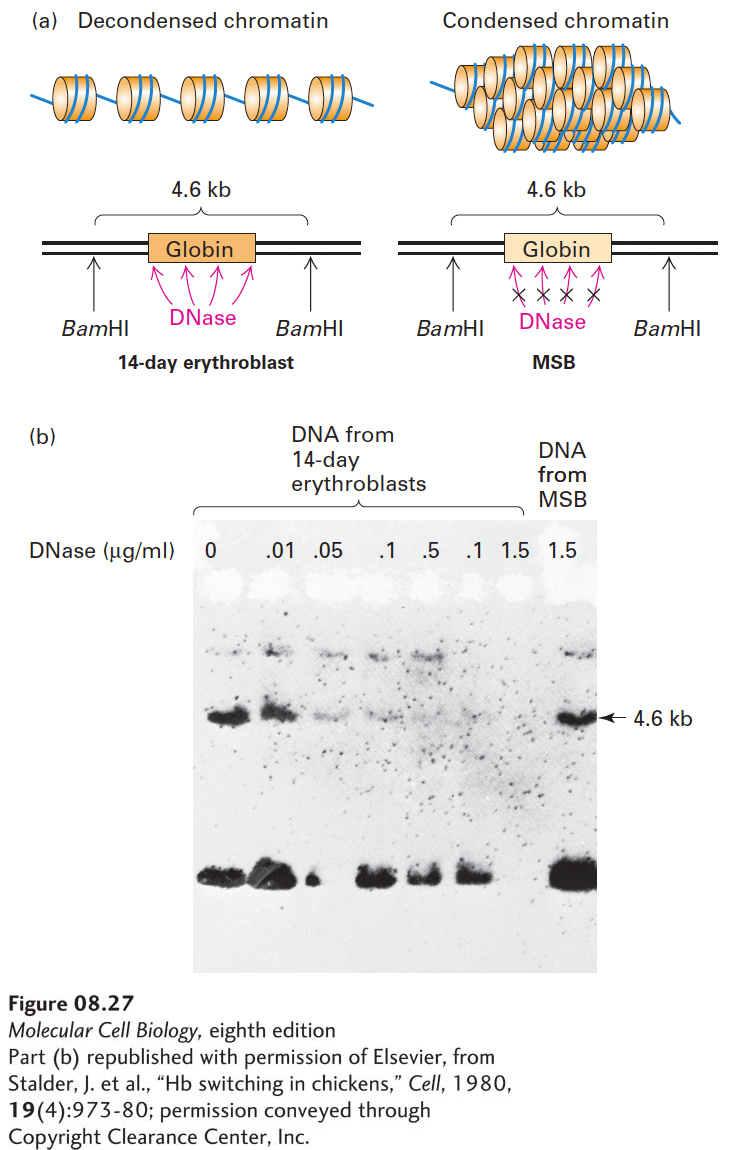
Histone acetylation at other sites in H4 and in other histones (see Figure 8-26b) is correlated with increased sensitivity of chromatin DNA to digestion by nucleases—an observation indicating that histone acetylation is associated with a less condensed form of chromatin. This correlation can be demonstrated by conducting experiments in which isolated nuclei are digested with DNase I. Following digestion, the DNA is completely separated from chromatin protein, digested to completion with a restriction enzyme, and analyzed by Southern blotting. An intact gene treated with a restriction enzyme yields fragments of characteristic sizes. If the nuclei are exposed first to DNase, however, the gene may be cleaved at random sites within the boundaries of the restriction enzyme cut sites. Consequently, any Southern blot bands normally seen with that gene will be lost. This method was first used to show that the β-globin gene in non-erythroid cells, in which it is transcriptionally inactive and in which it is associated with relatively nonacetylated histones, is much more resistant to DNase I than it is in erythroid progenitor cells, in which it is actively transcribed and in which it is associated with acetylated histones (Figure 8-27). These results indicate that the chromatin structure of nontranscribed DNA in hypoacetylated chromatin makes the DNA less accessible to DNase I than it is in transcribed, hyperacetylated chromatin. This is thought to be because chromatin containing the repressed gene is folded into condensed structures that sterically inhibit access to the associated DNA by the nuclease. In contrast, the transcribed gene is associated with a more unfolded form of chromatin, which allows better access of the nuclease to the associated DNA. Presumably, the condensed chromatin structure in non-erythroid cells also sterically inhibits access of the promoter and other transcription-control sequences in DNA to the proteins involved in transcription, contributing to transcriptional repression (see Chapter 9).
[Part (b) republished with permission of Elsevier, from Stalder, J. et al., “Hb switching in chickens,” Cell, 1980, 19(4):973-80; permission conveyed through Copyright Clearance Center, Inc.]
EXPERIMENTAL FIGURE 8-27Nontranscribed genes are less susceptible to DNase I digestion than active genes. Chick embryo erythroblasts at 14 days actively synthesize globin, whereas undifferentiated chicken lymphoblastic leukemia (MSB) cells do not. (a) Nuclei from each type of cell were isolated and exposed to increasing concentrations of DNase I. The nuclear DNA was then extracted and treated with the restriction enzyme BamHI, which cleaves the DNA around the globin sequence and normally releases a 4.6-kb globin fragment. (b) The DNase I- and BamHI-digested DNA was subjected to Southern blot analysis with a probe of labeled cloned adult globin DNA, which hybridizes to the 4.6-kb BamHI fragment. If the globin gene is susceptible to the initial DNase digestion, it should be cleaved repeatedly and would not be expected to show this fragment. As seen in the Southern blot, the transcriptionally active DNA from the 14-day globin-synthesizing cells was sensitive to DNase I digestion, indicated by the absence of the 4.6-kb band at high nuclease concentrations. In contrast, the inactive DNA from MSB cells was resistant to digestion. These results suggest that the inactive DNA is in a more condensed form of chromatin in which the globin gene is shielded from DNase digestion. See J. Stalder et al., 1980, Cell19:973.
[Part (b) republished with permission of Elsevier, from Stalder, J. et al., “Hb switching in chickens,” Cell, 1980, 19(4):973-80; permission conveyed through Copyright Clearance Center, Inc.]
Page 332
Genetic studies in yeast indicated that histone acetyl transferases (HATs), which acetylate specific lysine residues in histones, are required for the full activation of transcription of a number of genes. These enzymes are now known to have other substrates that influence gene expression in addition to histones. Consequently, they are more generally known as nuclear lysine acetyl transferases, or KATs, because K represents lysine in the single-letter code for amino acids (see Figure 2-14). Conversely, early genetic studies in yeast indicated that complete repression of many yeast genes requires the action of histone deacetylases (HDACs) that remove acetyl groups of acetylated lysines from histone tails, as discussed further in Chapter 9.
Other Histone Modifications As shown in Figure 8-26b, histone tails in chromatin can undergo a variety of other covalent modifications at specific amino acids. Lysine ε-amino groups can be methylated, a process that prevents acetylation, thus maintaining their positive charge. Moreover, the N of lysine ε-amino groups can be methylated once, twice, or three times. Arginine side chains can also be methylated. The O in hydroxyl groups (–OH) of serine and threonine side chains can be reversibly phosphorylated, introducing two negative charges. Each of these post-translational modifications contributes to the binding of chromatin-associated proteins that participate in the control of chromatin folding and the ability of DNA and RNA polymerases to replicate or transcribe the associated DNA. Finally, a single 76-amino-acid ubiquitin molecule can be reversibly added to a lysine in the C-terminal tails of H2A and H2B. Recall that addition of multiple linked ubiquitin molecules to a protein can mark it for degradation by the proteasome (see Figure 3-31). In this case, however, the addition of a single ubiquitin molecule does not affect the stability of a histone, although it does influence chromatin structure.
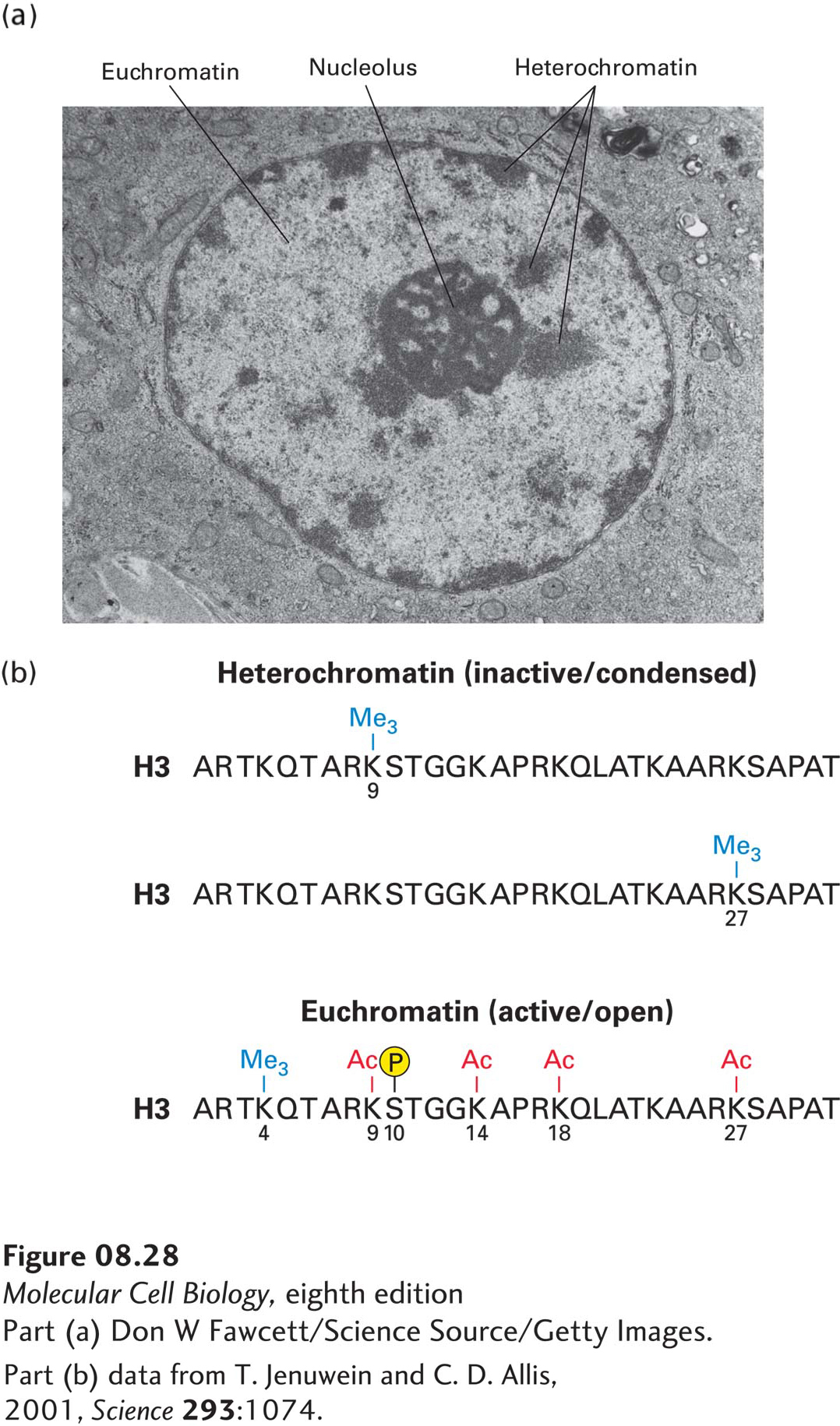
As mentioned previously, it is the precise combination of modified amino acids in histone tails that helps control the condensation, or compaction, of chromatin and its ability to be transcribed, replicated, and repaired. The extent of DNA compaction can be observed by electron microscopy and by light microscopy using dyes that bind DNA. Condensed regions of chromatin, known as heterochromatin, stain much more darkly than less condensed chromatin, known as euchromatin (Figure 8-28a). Heterochromatin does not fully decondense following mitosis, remaining in a compacted state during interphase and usually associating with the nuclear envelope, nucleoli, and additional distinct foci. Heterochromatin includes centromeres and telomeres of chromosomes as well as transcriptionally inactive genes. In contrast, areas of euchromatin, which are in a less compacted state during interphase, stain lightly with DNA dyes. Most transcribed regions of DNA are found in euchromatin. Heterochromatin usually contains histone H3 modified by methylation of lysine 9, while euchromatin generally contains histone H3 extensively acetylated on lysine 9 and other H3 lysines, especially at promoters and enhancers where there is also methylation of lysine 4, and phosphorylation of serine 10 (Figure 8-28b). Other histone tails are also differently modified in euchromatin and in heterochromatin. For example, H4 lysine 16 is generally nonacetylated in heterochromatin, which allows it to interact with neighboring nucleosomes and stabilize chromatin folding into the 30-nm fiber (see Figure 8-25).
[Part (a) Don W Fawcett/Science Source/Getty Images. Part (b) data from T. Jenuwein and C. D. Allis, 2001, Science293:1074.]
FIGURE 8-28Heterochromatin versus euchromatin. (a) In this electron micrograph of a bone marrow stem cell, the dark-staining areas in the nucleus (N) outside the nucleolus (n) are heterochromatin. The light-staining, whitish areas are euchromatin. (b) The modifications of histone N-terminal tails in heterochromatin and euchromatin differ, as illustrated here for histone H3. Note in particular that histone tails are generally much more extensively acetylated in euchromatin than in heterochromatin. Heterochromatin is much more condensed (thus less accessible to proteins) and is much less transcriptionally active than is euchromatin.
[Part (a) Don W Fawcett/Science Source/Getty Images. Part (b) data from T. Jenuwein and C. D. Allis, 2001, Science293:1074.]
Page 333
Reading the Histone Code The histone code of modified amino acids in the histone tails is “read” by proteins that bind to the modified tails and in turn promote condensation or decondensation of chromatin. Eukaryotes express a number of proteins containing a so-called chromodomain that binds to histone tails when they are methylated at specific lysines. One example of such a protein is heterochromatin protein 1 (HP1). The chromodomain of HP1 binds the H3 N-terminal tail only when it is di- or trimethylated at lysine 9 (see Figure 8-28b). HP1 also contains a second domain called a chromoshadow domain because it is frequently found in proteins that contain a chromodomain. The chromoshadow domain binds to other chromoshadow domains. Consequently, chromatin containing H3 di- and trimethylated at lysine 9 (H3K9Me2/3) is assembled into a condensed chromatin structure by HP1, although the structure of this chromatin is not well understood.
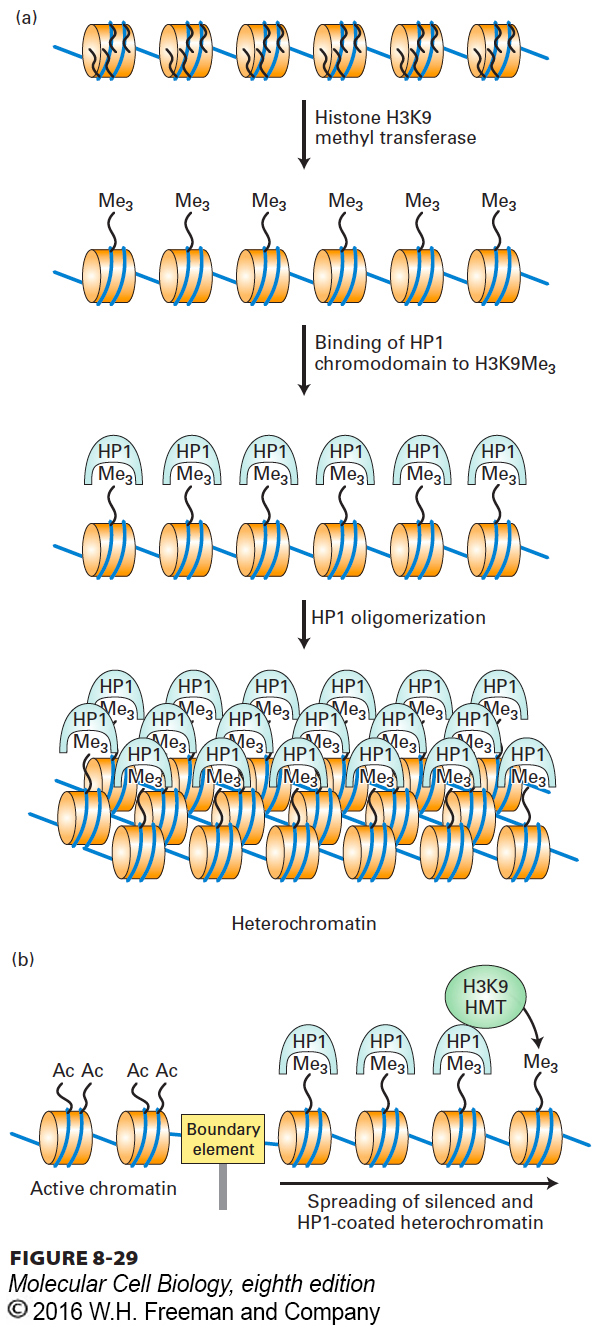
In addition to binding to itself, the chromoshadow domain of HP1 also binds the enzyme that methylates H3 lysine 9, an H3K9 histone methyl transferase (HMT) (Figure 8-29a). As a consequence, nucleosomes adjacent to a region of HP1-containing heterochromatin also become methylated at lysine 9 (Figure 8-29b). This methylation creates a binding site for another HP1 that can bind the H3K9 HMT, resulting in “spreading” of the heterochromatin structure along the chromosome until a boundary element is encountered that blocks further spreading. Boundary elements so far characterized are generally regions in chromatin where several nonhistone proteins bind to DNA, possibly blocking histone methylation on the other side of the boundary.
FIGURE 8-29Model for the formation of heterochromatin by the binding of HP1 to histone H3 trimethylated at lysine 9. (a) HP1 contributes to the condensation of heterochromatin by binding to histone H3 N-terminal tails trimethylated at lysine 9, then associating with other histone-bound HP1 molecules. (b) Heterochromatin condensation can spread along a chromosome because HP1 binds the histone methyltransferase (HMT) that methylates lysine 9 of histone H3. This creates a binding site for HP1 on the neighboring nucleosome. The spreading process continues until a “boundary element” is encountered. See G. Thiel et al., 2004, Eur. J. Biochem.271:2855, and A. J. Bannister et al., 2001, Nature410:120.
Epigenetic Memory Significantly, the model of heterochromatin formation in Figure 8-29b provides an explanation for how heterochromatic regions of a chromosome are reestablished following DNA replication during the S phase of the cell cycle. When DNA in heterochromatin is replicated, the histone octamers that are di- or trimethylated at H3 lysine 9 become distributed to both daughter chromosomes along with an equal number of newly assembled histone octamers. The H3K9 HMT associated with the H3K9 di- and trimethylated nucleosomes methylates lysine 9 of the newly assembled nucleosomes, regenerating the heterochromatin in both daughter chromosomes. Consequently, heterochromatin is marked with an epigenetic code, so called because it does not depend on the sequence of bases in DNA, that maintains the repression of associated genes in replicated daughter cells.
Other protein domains associate with histone-tail modifications typical of euchromatin. For example, the bromodomain binds to acetylated histone tails and therefore is associated with transcriptionally active chromatin. Several proteins involved in stimulating gene transcription contain bromodomains, such as the largest subunit of TFIID (see Chapter 9). This transcription factor contains two closely spaced bromodomains that probably help TFIID to associate with transcriptionally active chromatin (i.e., euchromatin). TFIID and other bromodomain-containing proteins also have histone acetylase activity, which helps to maintain the chromatin in a hyperacetylated state conducive to transcription. Consequently, an epigenetic code associated with euchromatin helps to maintain the transcriptional activity of genes in euchromatin through successive cell divisions. These epigenetic codes for heterochromatin and euchromatin help to maintain the patterns of gene expression established in different cell types during early embryonic development as specific differentiated cell types increase in numbers by cell division. Importantly, abnormal alterations in these epigenetic codes have been found to contribute to the pathogenic replication and behavior of cancer cells (see Chapter 24).
Page 334
In summary, multiple types of covalent modifications of histone tails can influence chromatin structure by altering nucleosome–nucleosome interactions and interactions with additional proteins that participate in or regulate processes such as transcription and DNA replication. The mechanisms and molecular processes governing chromatin modifications that regulate transcription are discussed in greater detail in the next chapter.
X-Chromosome Inactivation in Mammalian Females One important example of epigenetic gene control through repression by heterochromatin is the random inactivation and condensation of one of the two X chromosomes in female mammals. Each female mammal has two X chromosomes, one contributed by the egg from which it developed (Xm) and one contributed by the sperm (Xp). Early during embryonic development, random inactivation of either the Xm or the Xp chromosome occurs in each somatic cell. In the female embryo, about half the cells have an inactive Xm, and the other half have an inactive Xp. All subsequent daughter cells maintain the same inactive X chromosomes as their parent cells. As a result, the adult female is a mosaic of clones, some expressing the genes from the Xm and some expressing the genes from the Xp. This inactivation of one X chromosome in female mammals results in dosage compensation, a process that ensures that cells of females express proteins encoded on the X chromosome at the same levels as the cells of males, which have only one X chromosome.
Histones associated with the inactive X chromosome have post-translational modifications characteristic of other regions of heterochromatin: hypoacetylation of lysines, di- and trimethylation of histone H3 lysine 9, trimethylation of H3 lysine 27, and a lack of methylation at histone H3 lysine 4 (see Figure 8-28b). X-chromosome inactivation at an early stage in embryonic development is controlled by the X-inactivation center, a complex locus on the X chromosome that determines which of the two X chromosomes will be inactivated and in which cells. The X-inactivation center also contains the XIST gene, which encodes a remarkable long nonprotein-coding RNA that coats only the X chromosome it was transcribed from, thereby triggering silencing of that chromosome.
Although the mechanism of X-chromosome inactivation is not fully understood, it involves several processes, including the action of Polycomb protein complexes, which are discussed further in Chapter 9. One subunit of the Polycomb complex contains a chromodomain that binds to histone H3 tails when they are trimethylated at lysine 27. The Polycomb complex also contains a histone methyl transferase specific for H3 lysine 27. This finding helps to explain how the X-inactivation process spreads along large regions of the X chromosome and how it is maintained through DNA replication, apparently in a manner similar to heterochromatization by the binding of HP1 to histone H3 tails methylated at lysine 9 (see Figure 8-29b).
Page 335
X-chromosome inactivation is another example of an epigenetic process; that is, a process that affects the expression of specific genes and is inherited by daughter cells, but is not the result of a change in DNA sequence. In other words, the activity of genes on the X chromosome in female mammals is controlled by chromatin structure, rather than by the nucleotide sequence of the underlying DNA. And the inactivated X chromosome (either Xm or Xp) is maintained as the inactive chromosome in the progeny of all future cell divisions because the histones are modified in a specific, repressing manner that is faithfully inherited through each cell division.