26.5 Patterns of Evolution Are Revealed by Molecular Changes
The ability to analyze genetic variation at the molecular level has revealed a number of evolutionary processes and features that were formerly unsuspected. This section considers several aspects of evolution at the molecular level.
Rates of Molecular Evolution
Findings from molecular studies of numerous genes have demonstrated that different genes and different parts of the same gene often evolve at different rates.
Rates of Nucleotide Substitution
Rates of evolutionary change in nucleotide sequences are usually measured as the rate of nucleotide substitution, which is the number of substitutions taking place per nucleotide site per year within a population. To calculate the rate of nucleotide substitution, we begin by looking at homologous sequences from different organisms. We first align the homologous sequences and then compare the sequences and determine the number of nucleotides that differ between the two sequences. We might compare the growth-hormone sequences for mice and rats, which diverged from a common ancestor some 15 million years ago. From the number of different nucleotides in their growth-hormone genes, we compute the number of nucleotide substitutions that must have taken place since they diverged. Because the same site may have mutated more than once, the number of nucleotide substitutions is larger than the number of nucleotide differences in two sequences; special mathematical methods have been developed for inferring the actual number of substitutions likely to have taken place.
When we have the number of nucleotide substitutions per nucleotide site, we divide by the amount of evolutionary time that separates the two organisms (usually obtained from the fossil record) to obtain an overall rate of nucleotide substitution. For the mouse and rat growth-hormone gene, the overall rate of nucleotide substitution is approximately 8 × 10−9 substitutions per site per year.
Nonsynonymous and Synonymous Rates of Substitution
Nucleotide changes in a gene that alter the amino acid sequence of a protein are referred to as nonsynonymous substitutions. Nucleotide changes, particularly those at the third position of a codon, that do not alter the amino acid sequence, are called synonymous substitutions. The rate of nonsynonymous substitution varies widely among mammalian genes. The rate for the α-actin protein is only 0.01 × 10−9 substitutions per site per year, whereas the rate for interferon γ is 2.79 × 10−9, almost 300 times as high. The rate of synonymous substitution also varies among genes, but not to the extent of variation in the nonsynonymous rate. For most protein-encoding genes, the synonymous rate of change is considerably higher than the nonsynonymous rate because synonymous mutations are tolerated by natural selection (Table 26.4). Nonsynonymous mutations, on the other hand, alter the amino acid sequence of the protein and, in many cases, are detrimental to the fitness of the organism; most of these mutations are eliminated by natural selection.
| Gene | Nonsynonymous Rate (per site per 109 years) | Synonymous Rate (per site per 109 years) |
|---|---|---|
| α-Actin | 0.01 | 3.68 |
| β-Actin | 0.03 | 3.13 |
| Albumin | 0.91 | 6.63 |
| Aldolase A | 0.07 | 3.59 |
| Apoprotein E | 0.98 | 4.04 |
| Creatine kinase | 0.15 | 3.08 |
| Erythropoietin | 0.72 | 4.34 |
| α-Globin | 0.55 | 5.14 |
| β-Globin | 0.80 | 3.05 |
| Growth hormone | 1.23 | 4.95 |
| Histone 3 | 0.00 | 6.38 |
| Immunoglobulin heavy chain (variable region) | 1.07 | 5.66 |
| Insulin | 0.13 | 4.02 |
| Interferon α 1 | 1.41 | 3.53 |
| Interferon γ | 2.79 | 8.59 |
| Luteinizing hormone | 1.02 | 3.29 |
| Somatostatin-28 | 0.00 | 3.97 |
| Source: After W. Li and D. Graur, Fundamentals of Molecular Evolution (Sunderland, Mass.: Sinauer, 1991), p. 69, Table 1. | ||
Substitution Rates for Different Parts of a Gene
Different parts of a gene also evolve at different rates, with the highest rates of substitutions in regions of the gene that have the least effect on function, such as the third position of a codon, flanking regions, and introns (Figure 26.15). The 5′ and 3′ flanking regions of genes are not transcribed into RNA; therefore, substitutions in these regions do not alter the amino acid sequence of the protein, although they may affect gene expression (see Chapters 16 and 17). Rates of substitution in introns are nearly as high. Although these nucleotides do not encode amino acids, introns must be spliced out of the pre-mRNA for a functional protein to be produced, and particular sequences are required at the 5′ splice site, 3′ splice site, and branch point for correct splicing (see Chapter 14).
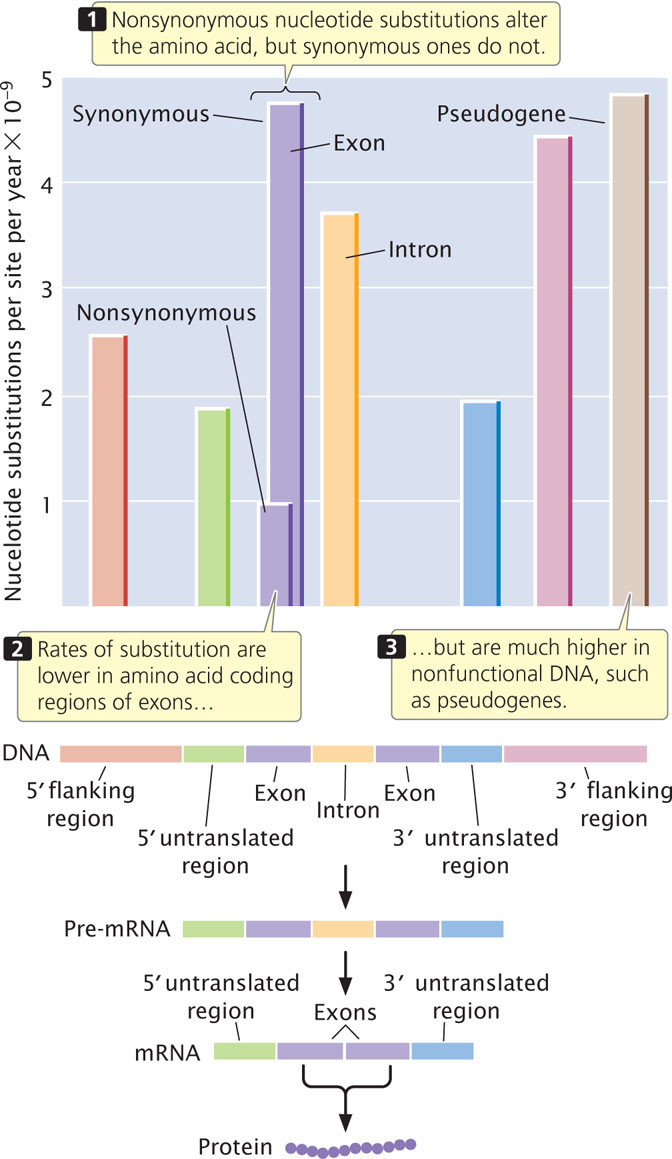
Substitution rates are somewhat lower in the 5′ and 3′ untranslated regions of a gene. These regions are transcribed into RNA but do not encode amino acids. The 5′ untranslated region contains the ribosome-binding site, which is essential for translation, and the 3′ untranslated region contains sequences that may function in regulating mRNA stability and translation; so substitutions in these regions may have deleterious effects on organismal fitness and may not be tolerated.
The lowest rates of substitution are seen in nonsynonymous changes in the coding region, because these substitutions always alter the amino acid sequence of the protein and are often deleterious. High rates of substitution occur in pseudogenes, most of which are duplicated nonfunctional copies of genes that have acquired mutations. Such genes usually no longer produce a functional product; so mutations in pseudogenes have little effect on the fitness of the organism.
In summary, there is a relation between the function of a sequence and its rate of evolution; higher rates are found where they have the least effect on function. This observation fits with the neutral-mutation hypothesis, which predicts that molecular variation is not affected by natural selection. ![]() TRY PROBLEM 30
TRY PROBLEM 30
The Molecular Clock
The neutral-mutation hypothesis proposes that evolutionary change at the molecular level takes place primarily through the fixation of neutral mutations by genetic drift. The rate at which one neutral mutation replaces another depends only on the mutation rate, which should be fairly constant for any particular gene. If the rate at which a protein evolves is roughly constant over time, the amount of molecular change that a protein has undergone can be used as a molecular clock to date evolutionary events.
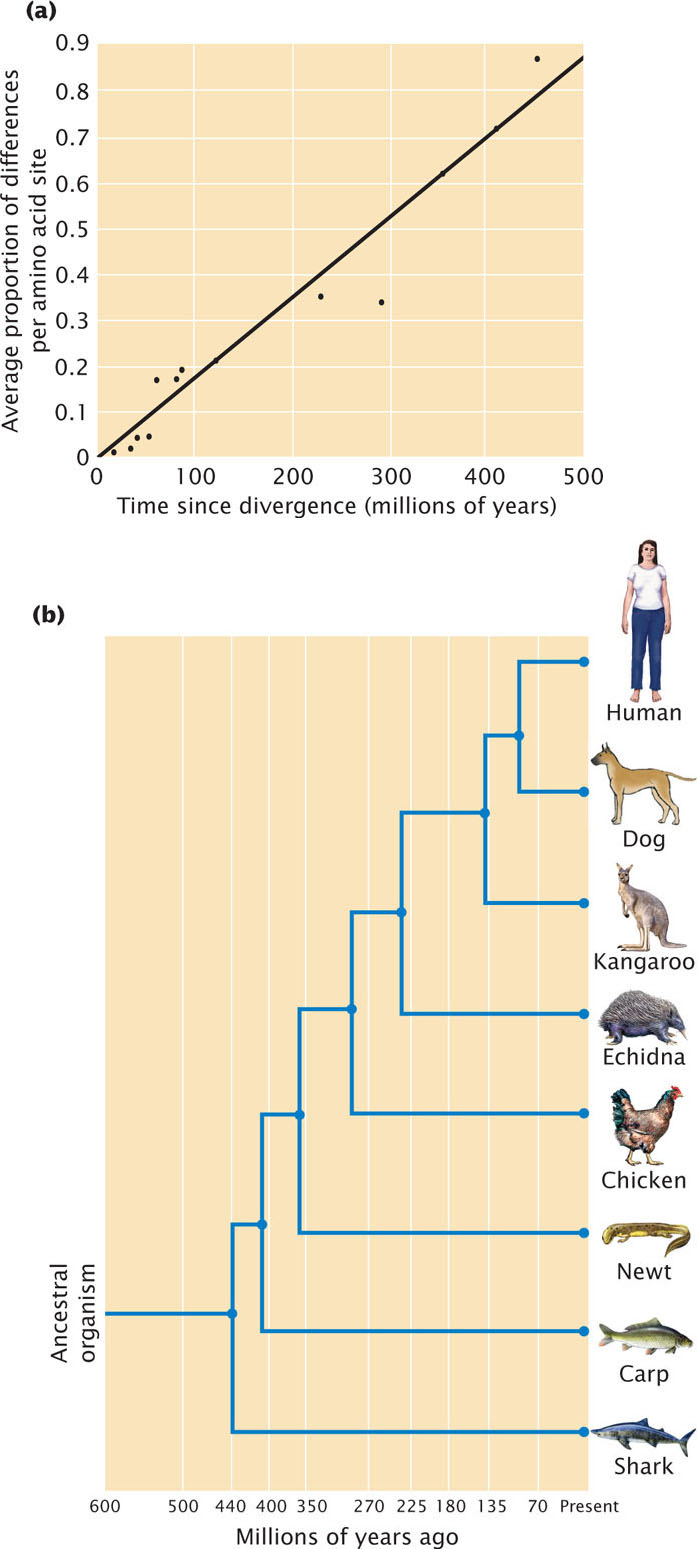
For example, the enzyme cytochrome c could be examined in two organisms known from fossil evidence to have had a common ancestor 400 million years ago. By determining the number of differences in the cytochrome c amino acid sequences in each organism, we could calculate the number of substitutions that have occurred per amino acid site. The occurrence of 20 amino acid substitutions since the two organisms diverged indicates an average rate of 5 substitutions per 100 million years. Knowing how fast the molecular clock ticks allows us to use molecular changes in cytochrome c to date other evolutionary events: if we found that cytochrome c in two organisms differed by 15 amino acid substitutions, our molecular clock would suggest that they diverged some 300 million years ago. If we assumed some error in our estimate of the rate of amino acid substitution, statistical analysis would show that the true divergence time might range from 160 million to 440 million years. The molecular clock was proposed by Emile Zuckerkandl and Linus Pauling in 1965 as a possible means of dating evolutionary events on the basis of molecules in present-day organisms. A number of studies have examined the rate of evolutionary change in proteins (Figure 26.16) and genes, and the molecular clock has been widely used to date evolutionary events when the fossil record is absent or ambiguous. For example, researchers used a molecular clock to estimate when Darwin’s finches diverged from a common ancestor that originally colonized the Galapagos Islands. This clock was based on DNA sequence differences in the cytochrome b gene. They concluded that the ancestor of Darwin’s finches arrived in the Galapagos and began diverging some 2-3 million years ago. The results of several studies have shown that the molecular clock does not always tick at a constant rate, particularly over shorter time periods, and this method remains controversial.
CONCEPTS
Different genes and different parts of the same gene evolve at different rates. Those parts of genes that have the least effect on function tend to evolve at the highest rates. The idea of the molecular clock is that individual proteins and genes evolve at a constant rate and that the differences in the sequences of present-day organisms can be used to date past evolutionary events.
 CONCEPT CHECK 7
CONCEPT CHECK 7In general, which types of sequences are expected to exhibit the slowest evolutionary change?
- Synonymous changes in amino acid coding regions of exons.
- Nonsynonymous changes in amino acid coding regions of exons.
- Introns.
- Pseudogenes.
Evolution Through Changes in Gene Regulation
One of the challenges of evolutionary biology is understanding the genetic basis of adaptation. Many evolutionary changes occur with relatively few genetic differences. For example, humans and chimpanzees differ greatly in anatomy, physiology, and behavior and yet differ at only about 4% of their DNA sequences (see introduction to Chapter 17). Evolutionary biologists have long assumed that many anatomical differences result, not from the evolution of new genes, but rather from relatively small DNA differences that alter the expression of existing genes. Recent research in evolutionary genetics has focused on how evolution occurs through alteration of gene expression.
An example of adaptation that has occurred through changes to regulatory sequences is seen in the evolution of pigmentation in Drosophila melanogaster fruit flies in Africa. Most fruit flies are light tan in color, but flies in some populations of Africa exhibit much darker abdomens. These darker flies usually occur in mountainous regions at higher elevations. Indeed, 59% of pigmentation variation among populations within sub-Saharan Africa can be explained by differences in elevation (Figure 26.17). Researchers have demonstrated that these differences are genetically determined and that natural selection has favored darker pigmentation at high elevation. High-elevation populations are exposed to lower temperatures, and the darker pigmentation is assumed to help flies absorb more solar radiation and better regulate their body temperature in these environments.
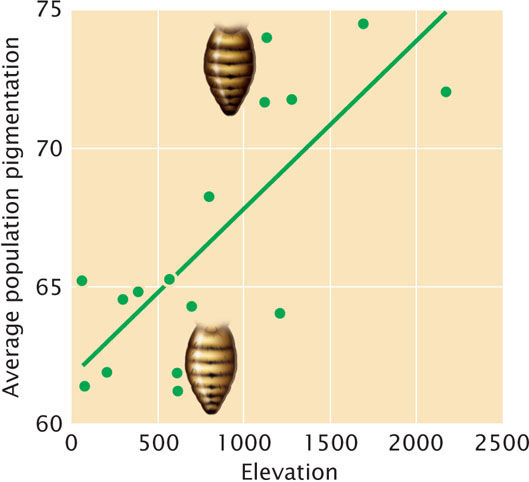
How did flies at high elevation evolve darker color? Genetic studies indicate that the dark abdominal pigmentation seen in flies from these populations results from variation at or near a locus called ebony. The ebony locus encodes a multifunctional enzyme that produces an exoskeleton which is yellow in color; its absence produces a dark phenotype. Sequencing of the ebony locus of flies from light and dark populations found no differences in the coding region of the ebony gene. However, molecular analysis revealed a marked reduction in the amount of ebony mRNA in darker flies, suggesting that the differences in pigmentation are not due to mutations at the ebony gene itself, but rather in its expression. Further investigation detected genetic differences within an enhancer that is about 3,600 bp upstream of the ebony gene. Dark and light flies differed in over 120 nucleotides scattered over 2,400 bp of the enhancer. However, by experimentally creating enhancers with different combinations of these mutations, researchers determined that five of the mutations are responsible for the majority of the differences in pigmentation.
These studies suggested that over time high-elevation populations accumulated multiple mutations in the enhancer, which reduced the expression of the ebony locus and caused darker pigmentation. Further analysis suggested that these mutations were added sequentially. Some of the mutations are widespread throughout Africa; it is assumed that these existing mutations were favored by natural selection in high-elevation populations and increased in frequency because they helped the flies thermoregulate in colder environments. Other mutations are only seen in the high-elevation populations, suggesting that they probably arose as new mutations within these populations and were quickly favored by natural selection.
Genome Evolution
The rapid growth of sequence data available in DNA databases has been a source of insight into evolutionary processes. Whole-genome sequences also are providing new information about how genomes evolve and the processes that shape the size, complexity, and organization of genomes.
Exon Shuffling
Many proteins are composed of groups of amino acids, called domains, that specify discrete functions or contribute to the molecular structure of a protein. For example, in Chapter 16, we considered the DNA-binding domains of proteins that regulate gene expression. Analyses of gene sequences from eukaryotic organisms indicate that exons often encode discrete functional domains of proteins.
Some genes elongated and evolved new functions when one or more exons duplicated and underwent divergence. For example, the human serum-albumin gene is made up of three copies of a sequence that encodes a protein domain consisting of 195 amino acids. Additionally, the genes that encode human immunoglobulins have undergone repeated tandem duplications, creating many similar V, J, D, and C segments that enable the immune system to respond to almost any foreign substance that enters the body.
A comparison of DNA sequences from different genes reveals that new genes have repeatedly evolved through a process called exon shuffling, in which exons of different genes are exchanged, creating genes that are mosaics of other genes. For example, tissue plasminogen activator (TPA) is an enzyme that contains four domains of three different types, called kringle, growth factor, and finger. Each domain is encoded by a different exon. The gene for TPA is believed to have acquired its exons from other genes that encode different proteins: the kringle exon came from the plasminogen gene, the growth-factor exon came from the epidermal growth-factor gene; and the finger exon came from the fibronectin gene. The mechanism by which exon shuffling takes place is poorly known, but new proteins with different combinations of functions encoded by other genes apparently have repeatedly evolved by this mechanism.
Gene Duplication
New genes have also evolved through the duplication of whole genes and their subsequent divergence. This process creates multigene families, sets of genes that are similar in sequence but encode different products. For example, humans possess 13 different genes found on chromosomes 11 and 16 that encode globinlike molecules, which take part in oxygen transport (Figure 26.18). All of these genes have a similar structure, with three exons separated by two introns, and are assumed to have evolved through repeated duplication and divergence from a single globin gene in a distant ancestor. This ancestral gene is thought to have been most similar to the present-day myoglobin gene and first duplicated to produce an α/β-globin precursor gene and the myoglobin gene. The α/β-globin gene then underwent another duplication to give rise to a primordial α-globin gene and a primordial β-globin gene. Subsequent duplications led to multiple α-globin and β-globin genes. Similarly, vertebrates contain four clusters of Hox genes, each cluster comprising from 9 to 11 genes. Hox genes play an important role in development.
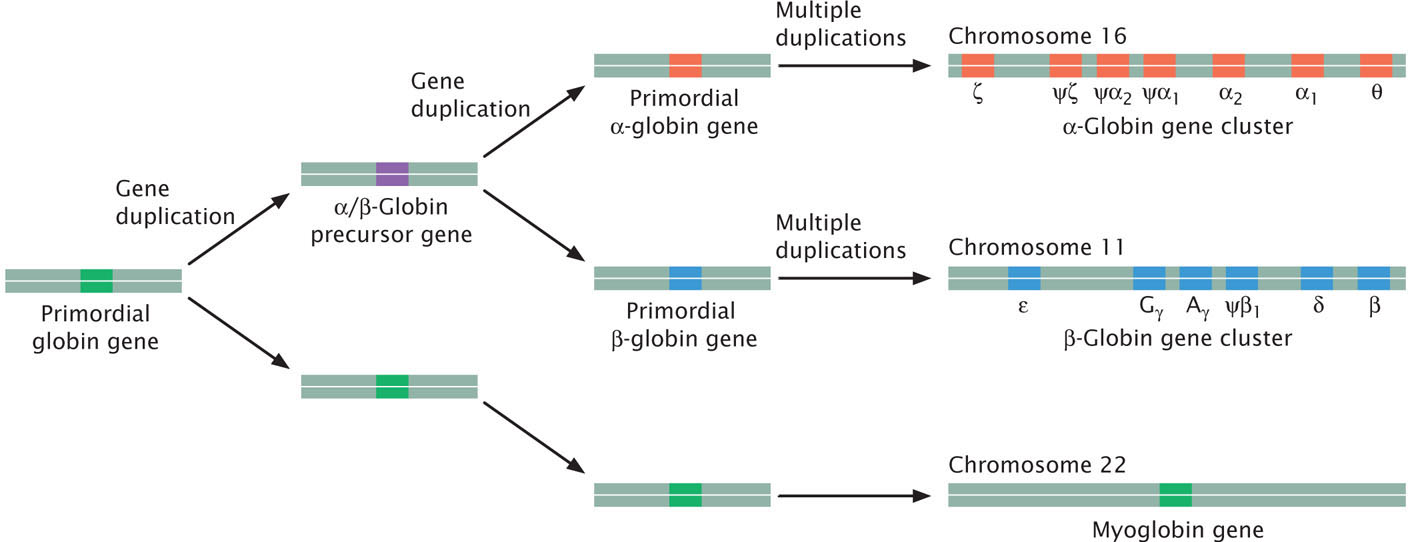
Some gene families include genes that are arrayed in tandem on the same chromosome; others are dispersed among different chromosomes. Gene duplication is a common occurrence in eukaryotic genomes; for example, about 5% of the human genome consists of duplicated segments.
Gene duplication provides a mechanism for the addition of new genes with novel functions; after a gene duplicates, there are two copies of the sequence, one of which is free to change and potentially take on a new function. The extra copy of the gene may, for example, become active at a different time in development or be expressed in a different tissue or even diverge and encode a protein having different amino acids. However, the most common fate of gene duplication is that one copy acquires a mutation that renders it nonfunctional, giving rise to a pseudogene. Pseudogenes are common in the genomes of complex eukaryotes; the human genome is estimated to contain as many as 20,000 pseudogenes.
Whole-Genome Duplication
In addition to the duplication of individual genes, whole genomes of some organisms have duplicated in the past. For example, a comparison of the genome of the yeast Saccharomyces cerevisiae with the genomes of other fungi reveals that S. cerevisiae or one of its immediate ancestors underwent a whole-genome duplication, generating two copies of every gene. Many of the copies subsequently acquired new functions; others acquired mutations that destroyed the original function and then diverged into random DNA sequences. Whole-genome duplication can take place through polyploidy.
During their evolution, plants have undergone a number of whole-genome duplications through polyploidy. While polyploidy is less common in animals, genetic evidence suggests that several whole-genome duplication events have occurred during animal evolution. In 1970, Susumu Ohno proposed that early vertebrates underwent two rounds of genome duplication. Called the 2R hypothesis, this idea has been controversial, but recent data from genome sequencing has provided support for it.
Horizontal Gene Transfer
Traditionally, scientists assumed that organisms acquire their genomes through vertical transmission—transfer through the reproduction of genetic information from parents to offspring, and most phylogenetic trees assume vertical transmission of genetic information. Findings from DNA sequence studies reveal that DNA sequences are sometimes exchanged by a horizontal gene transfer, in which DNA is transferred between individuals of different species (see Chapter 9). This process is especially common among bacteria, and there are a number of documented cases in which genes are transferred from bacteria to eukaryotes. The extent of horizontal gene transfer among eukaryotic organisms is controversial, with few well-documented cases. Horizontal gene transfer can obscure phylogenetic relationships and make the reconstruction of phylogenetic trees difficult.
One apparent case of horizontal gene transfer among eukaryotes is the presence in some aphids of genes for enzymes that synthesize carotenoids. Carotenoids are colored compounds produced by bacteria, archaea, fungi, and plants. Many animals also have carotenoids, but they lack the enzymes necessary of make the compounds themselves; in almost all cases, animals obtain carotenoids from their food.
Aphids—small insects that feed on plants—have carotenoids, which are responsible for color differences between and within species. Some aphids are green and contain α-, β-, and γ-carotene, which are all yellow carotenoids. Other aphids are red or brown and contain lycopene or torulene, carotenoids that are red. One species, the pea aphid (Acyr-thosiphon pisum) has both green and red individuals, and these differences are genetically inherited. Many researchers previously assumed that the color differences were due to carotenoids that were acquired in the aphids’ food.
Researchers have recently sequenced the entire genome of A. pisum, which provided the opportunity to determine whether the aphids possessed their own genes for carotenoid synthesis. Examination of genomic sequences revealed that pea aphids have several genes that code for carotenoid-synthesizing enzymes. Interestingly, these genes are closely related to carotenoid-synthesizing genes found in some fungi. The evidence suggests that in the distant past, an aphid acquired its carotenoid genes from a fungus through horizontal gene transfer and then passed the genes on to other aphids through vertical transmission.
CONCEPTS
New genes may evolve through the duplication of exons, shuffling of exons, duplication of genes, and duplication of whole genomes. Genes can be passed among distantly related organisms through horizontal gene transfer.