Types of Gene Mutations
There are a number of ways to classify gene mutations. Some classification schemes are based on the nature of the phenotypic effect, others are based on the causative agent of the mutation, and still others focus on the molecular nature of the defect. Here, we will categorize mutations primarily on the basis of their molecular nature, but we will also encounter some terms that relate to the causes and the phenotypic effects of mutations.
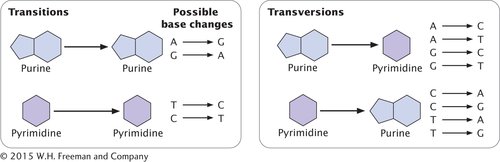
BASE SUBSTITUTIONS The simplest type of gene mutation is a base substitution, the alteration of a single nucleotide in the DNA (Figure 13.2a). There are two types of base substitutions. In a transition, a purine is replaced by a different purine or, alternatively, a pyrimidine is replaced by a different pyrimidine (Figure 13.3). In a transversion, a purine is replaced by a pyrimidine or a pyrimidine is replaced by a purine. The number of possible transversions (see Figure 13.3) is twice the number of possible transitions, but transitions arise more frequently because transforming a purine into different purine or a pyrimidine into different pyrimidine is easier than transforming a purine into pyrimidine, or vice versa.  TRY PROBLEM 13
TRY PROBLEM 13
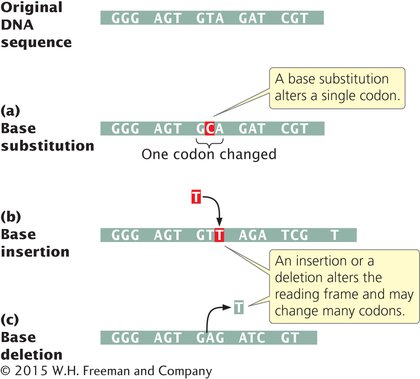
INSERTIONS AND DELETIONS Another class of gene mutations contains insertions and deletions (collectively called indels): the addition or removal, respectively, of one or more nucleotide pairs (Figure 13.2b and c). Although base substitutions are often assumed to be the most common type of mutation, molecular analysis has revealed that insertions and deletions are frequently more common. Insertions and deletions within sequences that encode proteins may lead to frameshift mutations: changes in the reading frame (Chapter 11) of the gene. Frameshift mutations usually alter all amino acids encoded by the nucleotides following the mutation, so they generally have drastic effects on the phenotype. Some frameshifts also introduce premature stop codons, terminating protein synthesis early and resulting in a shortened (truncated) protein. Not all insertions and deletions lead to frameshifts, however; insertions and deletions consisting of any multiple of three nucleotides leave the reading frame intact, although the addition or removal of one or more amino acids may still affect the phenotype. Indels that do not affect the reading frame are called in-
CONCEPTS
Gene mutations consist of changes in a single gene and can be base substitutions (a single pair of nucleotides is altered) or insertions or deletions (nucleotides are added or removed). A base substitution can be a transition (substitution of like bases) or a transversion (substitution of unlike bases). Insertions and deletions often lead to a change in the reading frame of a gene.
 CONCEPT CHECK 1
CONCEPT CHECK 1
Which of the following changes is a transition base substitution?
Adenine is replaced by thymine.
Cytosine is replaced by adenine.
Guanine is replaced by adenine.
Three nucleotide pairs are inserted into DNA.
c
EXPANDING NUCLEOTIDE REPEATS Mutations in which the number of copies of a set of nucleotides increases are called expanding nucleotide repeats. This type of mutation was first observed in 1991 in a gene called FMR-
Expanding nucleotide repeats have been found in a number of other human genetic diseases, several of which are listed in Table 13.1. Most of these diseases are caused by the expansion of a set of three nucleotides (called a trinucleotide), most often CNG, where N can be any nucleotide. However, some diseases are caused by repeats of four, five, and even twelve nucleotides. The number of copies of the nucleotide repeat often correlates with the severity or age of onset of the disease. The number of copies of the repeat also corresponds to its instability: when more repeats are present, the probability of expansion to even more repeats increases.
| Number of copies of repeat | |||
|---|---|---|---|
| Disease | Repeated sequence | Normal range | Disease range |
| Spinal and bulbar muscular atrophy | CAG | 11–33 | 40–62 |
| Fragile- |
CGG | 6–54 | 50–1500 |
| Jacobsen syndrome | CGG | 11 | 100–1000 |
| Spinocerebellar ataxia (several types) | CAG | 4–44 | 21–130 |
| Autosomal dominant cerebellar ataxia | CAG | 7–19 | 37–220 |
| Myotonic dystrophy | CTG | 5–37 | 44–3000 |
| Huntington disease | CAG | 9–37 | 37–121 |
| Friedreich ataxia | GAA | 6–29 | 200–900 |
| Dentatorubral- |
CAG | 7–25 | 49–75 |
| Myoclonus epilepsy of the Unverricht–Lundborg type | CCCCGCCCCGCG | 2–3 | 12–13 |
Increases in the number of nucleotide repeats can produce disease symptoms in different ways. In several diseases (e.g., Huntington disease), the nucleotide expansion occurs within the coding part of a gene, producing a toxic protein that has extra glutamine residues (the amino acid encoded by CAG). In other diseases, the repeat is outside the coding region of a gene and affects its expression. In fragile-
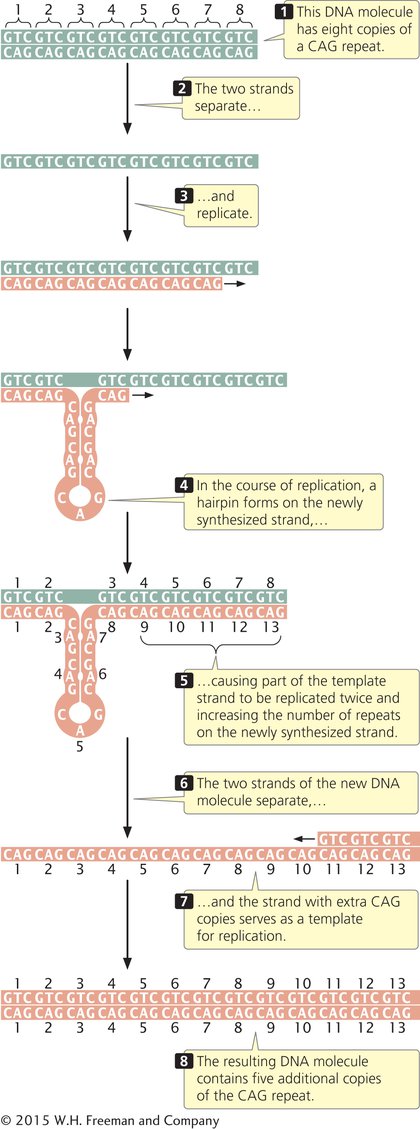
A possible source of nucleotide repeat expansion is the formation of hairpins and other special DNA structures, which can cause nucleotides in the template strand to be replicated twice, thus increasing the number of repeats on the newly synthesized strand (Figure 13.5). Watching Animation 13.1 will help you understand how the nucleotide repeats increase in number.
CONCEPTS
Expanding nucleotide repeats are regions of DNA that consist of repeated copies of sets of nucleotides. Increased numbers of nucleotide repeats are associated with several genetic diseases.