By 1980, methods for mapping and sequencing DNA fragments had been sufficiently developed that geneticists began seriously proposing that the entire human genome be sequenced. An international collaboration was planned to undertake the Human Genome Project; initial estimates suggested that 15 years and $3 billion would be required to accomplish the task.
The Human Genome Project officially began in October 1990. Initial efforts focused on developing new and automated methods for cloning and sequencing DNA and on generating detailed physical and genetic maps of the human genome. By 1993, large-scale physical maps were completed for all 23 pairs of human chromosomes. At the same time, automated sequencing techniques had been developed that made large-scale sequencing feasible.
The initial effort to sequence the genome was a public project consisting of an international collaboration among 20 research groups and hundreds of individual researchers who formed the International Human Genome Sequencing Consortium. This group used a map-based strategy for sequencing the human genome.
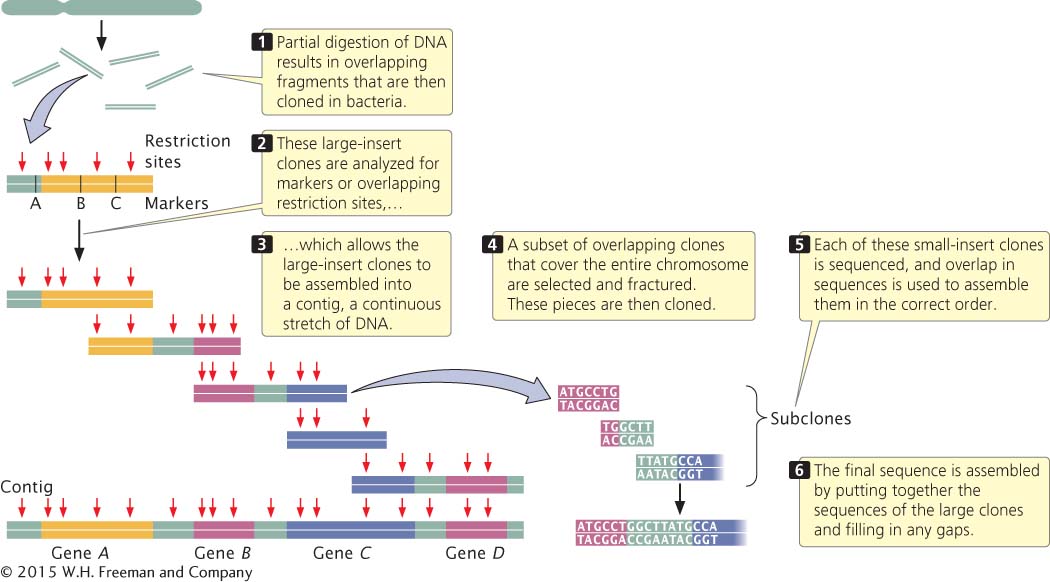
MAP-BASED SEQUENCING In map-based sequencing, short sequenced fragments are assembled into a whole-genome sequence by first creating detailed genetic and physical maps of the genome, which provide known locations of genetic markers (restriction sites, known genes, or known DNA sequences) at regularly spaced intervals along each chromosome. These markers are later used to help align the short sequenced fragments into their correct order.
After the genetic and physical maps are available, chromosomes or large pieces of chromosomes are separated by flow cytometry, in which chromosomes are sorted optically by size, or by pulsed-field gel electrophoresis, in which large molecules are separated in a gel by alternating the direction of the current. Each chromosome (or sometimes the entire genome) is then cut up by partial digestion with restriction enzymes (Figure 15.3). Partial digestion means that the restriction enzymes are allowed to act for only a limited time so that not all restriction sites in every DNA molecule are cut. Thus, partial digestion produces a set of large overlapping DNA fragments, which are then cloned with the use of cosmids, yeast artificial chromosomes (YACs), or bacterial artificial chromosomes (BACs).
15.3 Map-based approaches to whole-genome sequencing rely on detailed genetic and physical maps to align sequenced fragments.
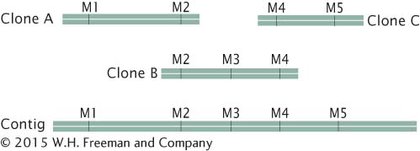
Next, these large-insert clones are put together in their correct order on the chromosome (see Figure 15.3). This assembly can be done in several ways. One method relies on the presence of a high-density map of genetic markers. A complementary DNA probe is made for each genetic marker, and a library of the large-insert clones is screened with the probe, which hybridizes to any colony containing a clone with the marker. The library is then screened for neighboring markers. Because the clones are much larger than the markers used as probes, some clones will have more than one marker. For example, clone A might have markers M1 and M2, clone B markers M2, M3, and M4, and clone C markers M4 and M5. Such a result would indicate that these clones contain areas of overlap, as shown here:
A set of two or more overlapping DNA fragments that form a contiguous stretch of DNA is called a contig. This approach was used in 1993 to create a contig consisting of 196 overlapping YAC clones (see Figure 15.2) of the human Y chromosome.
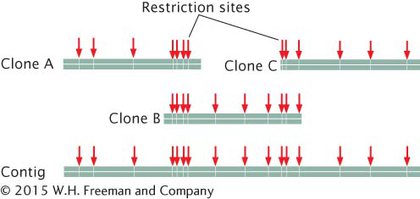
The order of clones can also be determined without the use of preexisting genetic maps. For example, each clone can be cut with a series of restriction enzymes and the resulting fragments then separated by gel electrophoresis. This method generates a unique set of restriction fragments, called a fingerprint, for each clone. The restriction patterns for the clones are stored in a database. A computer program is then used to examine the restriction patterns of all the clones and look for areas of overlap. The overlap is then used to arrange the clones in order, as shown here:
Page 407
Other genetic markers can be used to help position contigs along the chromosome.
When the large-insert clones have been assembled into the correct order on the chromosome, a subset of overlapping clones that efficiently cover the entire chromosome can be chosen for sequencing; the goal is to select the minimum number of clones that is necessary to represent the chromosome. Each of the selected large-insert clones is fractured into smaller overlapping fragments, which are themselves cloned (see Figure 15.3). These smaller clones (called small-insert clones or subclones) are then sequenced. The sequences of the small-insert clones are examined for overlap, which allows them to be correctly assembled to give the sequence of the large-insert clones. Enough overlapping small-insert clones are usually sequenced to ensure that the entire genome is sequenced several times. Finally, the whole genome is assembled by putting together the sequences of all overlapping contigs. Often, gaps in the genome sequence still exist and must be filled in by using other methods.
The International Human Genome Sequencing Consortium used a map-based approach to sequencing the human genome. Many copies of the human genome were cut up into fragments of about 150,000 bp each, which were inserted into bacterial artificial chromosomes. Restriction fingerprints were used to assemble the BAC clones into contigs, which were positioned on the chromosomes with the use of genetic markers and probes. The individual BAC clones were sheared into smaller overlapping fragments and sequenced, and the whole genome was assembled by putting together the sequence of the BAC clones.
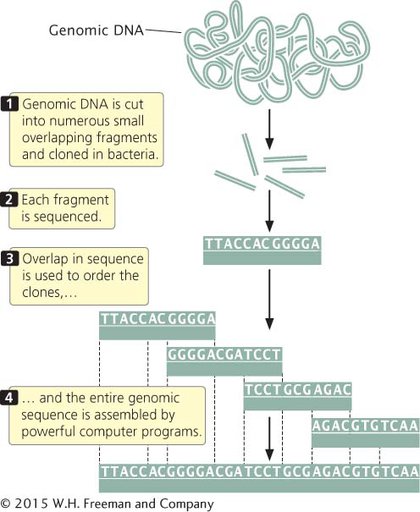
WHOLE-GENOME SHOTGUN SEQUENCING In 1998, Craig Venter announced that he would lead a company called Celera Genomics in a private effort to sequence the human genome. He proposed using a shotgun sequencing approach, which he suggested would be quicker than the map-based approach employed by the International Human Genome Sequencing Consortium. In whole-genome shotgun sequencing (Figure 15.4), the entire genome is assembled based on sequence overlap. Small-insert clones are prepared directly from genomic DNA and sequenced. Powerful computer programs then assemble the entire genome by examining overlap in the nucleotide sequences among the small-insert clones. One advantage of shotgun sequencing is that the small-insert clones can be placed in plasmids, which are simple and easy to manipulate. The requirement for overlap means that most of the genome must be sequenced multiple (often 10 to 15) times. The average number of times a nucleotide in the genome is sequenced is called the sequencing coverage. For example, 10× coverage means that an average nucleotide in the genome has been sequenced 10 times.
Shotgun sequencing was initially used for assembling small genomes such as those of bacteria. When Venter proposed the use of this approach for sequencing the human genome, it was not at all clear that the approach could successfully assemble a complex genome consisting of billions of base pairs. Today, virtually all genomes are sequenced using the whole-genome shotgun approach. TRY PROBLEM 15
CONCEPTS
Sequencing a genome requires breaking it up into small overlapping fragments whose DNA sequences can be determined in a sequencing reaction. In map-based sequencing, sequenced fragments are ordered into the final genome sequence with the use of genetic and physical maps. In whole-genome shotgun sequencing, the genome is assembled by means of overlap in the sequences of small fragments.
CONCEPT CHECK 1
A contig is
a set of molecular markers used in genetic mapping.
a set of overlapping fragments that form a continuous stretch of DNA.
a set of fragments generated by a restriction enzyme.
a small DNA fragment used in sequencing.
b
For several years, the public effort by the International Human Genome Sequencing Consortium, using a map-based approach, and the private Celera effort, using shotgun sequencing, moved forward simultaneously. In the summer of 2000, both public and private sequencing projects announced the completion of a rough draft that included most of the sequence of the human genome, five years ahead of schedule (Figure 15.5). An analysis of this sequence was published 6 months later. The human genome sequence was declared completed in the spring of 2003, although some gaps still remain. For most chromosomes, the finished sequence is 99.999% accurate, with less than one error per 100,000 bp, an accuracy rate 10 times that of the initial goal.
15.5 Craig Venter (left), president of Celera Genomics, and Francis Collins (right), director of the National Human Genome Research Institute, NIH, announced the completion of a rough draft of the human genome at a press conference in Washington, D.C., on June 26, 2000.
[Alex Wong/Newsmakers/Getty Images.]
RESULTS AND IMPLICATIONS OF THE HUMAN GENOME PROJECT With the first human genome determined, sequencing individual genomes is much easier. It is now possible to sequence an entire human genome in a single day, and genomes from thousands of different people have now been completely sequenced. The cost of sequencing a complete human genome has also dropped dramatically and will continue to fall as sequencing technology improves.
The availability of the complete sequence of the human genome is proving to be of great benefit. The sequence has provided tools for detecting and mapping genetic variants across the human genome, greatly facilitating gene mapping in humans. For example, several million sites at which people differ in a single nucleotide (called single-nucleotide polymorphisms; see Section 15.2) have now been identified, and these sites are being used in genome-wide association studies to locate genes that affect diseases and traits in humans (see Chapter 5). The sequence is also providing important information about development and many basic cellular processes.
Page 409
Next-generation sequencing techniques that allow rapid and inexpensive sequencing of genomic DNA (see Chapter 14) are being used to address fundamental questions in many areas. For example, the genomes of a number of cancer cells have now been completely sequenced and compared with the sequence of healthy cells from the same person, allowing complete determination of all the mutations that lead to tumor formation and cancer progression. The complete genome of an unborn baby has been sequenced from fetal DNA isolated from its mother’s blood. The 1000 Genomes Project is sequencing and comparing the genomes of several thousand people from different ethnic groups, with the goal of detecting most of the common variations that exist in the human species. Sequencing of the complete genomes of parents and their children has allowed a direct estimate of mutation rates.
DNA has been extracted from the bones of ancient humans, including Neanderthals and Denisovans (a little-known group of humans that appear to be closely related to Neanderthals), and completely sequenced. Comparisons of the modern human genome with the sequences of these and other species are adding to our understanding of human evolution as well as our knowledge of the evolution and the history of all life.
In spite of these benefits and successes, some people have been disappointed by the lack of tangible results from the Human Genome Project. At the time it was proposed, there was speculation that the sequencing of the human genome would immediately revolutionize the practice of medicine, leading to new insights for treating common diseases and resulting in the development of powerful new drugs. Although genome sequence data have produced numerous new and exciting research findings and have led to a better understanding of many diseases, the data are still seldom used by practicing physicians in the treatment of patients. Undoubtedly, genomic information will be important for medicine in the future, both for tailoring treatment to individual patients (personalized medicine) and for drug discovery, but when this will take place is currently uncertain.
Along with the many potential benefits of complete genome sequence information, there are concerns about its misuse. With the knowledge gained from genome sequencing, many more genes for diseases, disorders, and behavioral and physical traits will be identified, increasing the number of genetic tests that can be performed to make predictions about the future phenotype and health of a person. There is concern that information from genetic testing might be used to discriminate against people who are carriers of disease-causing genes or who might be at risk for some future disease. This problem has been addressed to some extent in the United States with the passage of the Genetic Information Nondiscrimination Act, which prohibits health insurers and employers from using genetic information to make decisions about health insurance coverage and employment (although it does not apply to life, disability, or long-term care insurance). Questions also arise about who should have access to a person’s genome sequence. What about relatives, who have similar genomes and might also be at risk for some of the same diseases? There are also questions about the use of this information to select for specific traits in future offspring. All of these concerns are legitimate and must be addressed if we are to use the information from genome sequencing responsibly.
CONCEPTS
The Human Genome Project was an effort to sequence the entire human genome. Begun in 1990, the effort was undertaken by two competing teams, an international consortium of publicly supported investigators and a private company, both of which finished a rough draft of the genome sequence in 2000. The entire sequence was completed in 2003. The ability to rapidly sequence human genomes raises a number of ethical questions.
 TRY PROBLEM 15
TRY PROBLEM 15 CONCEPT CHECK 1
CONCEPT CHECK 1