CONCEPT15.5 Genomes Reveal Both Neutral and Selective Processes of Evolution
Most natural populations harbor far more genetic variation than we would expect to find if genetic variation were influenced by natural selection alone. This discovery, combined with the knowledge that many mutations do not change molecular function, provided a major stimulus to the development of the field of molecular evolution.
To discuss the evolution of genes, we need to consider the specific types of mutations that are possible. A nucleotide substitution is a change in a single nucleotide in a DNA sequence (a type of point mutation). Many nucleotide substitutions have no effect on phenotype, even if the change occurs in a gene that encodes a protein, because most amino acids are specified by more than one codon. A substitution that does not change the encoded amino acid is known as a synonymous substitution (also called a silent substitution; FIGURE 15.17A). Synonymous substitutions do not affect the functioning of a protein (although they may have other effects, such as changes in mRNA stability or translation rates) and are therefore less likely to be influenced by natural selection.
313
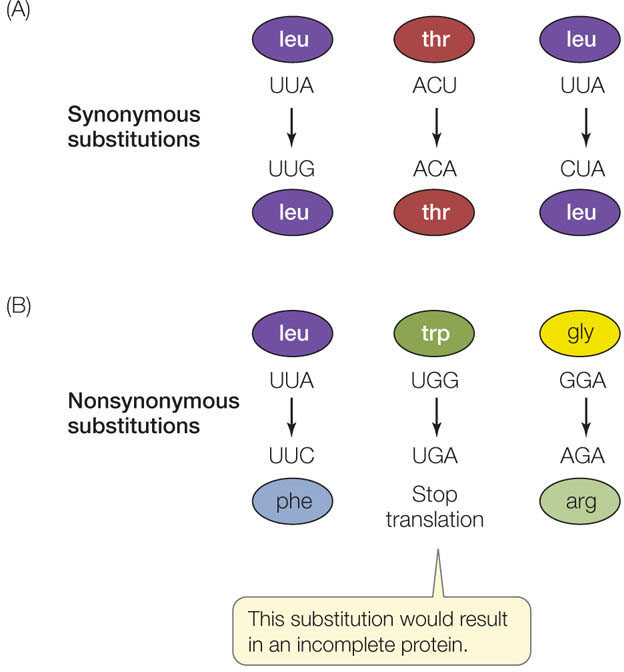
A nucleotide substitution that does change the amino acid sequence encoded by a gene is known as a nonsynonymous substitution (also called a missense substitution; FIGURE 15.17B). In general, nonsynonymous substitutions are likely to be deleterious to the organism. But not every amino acid replacement alters a protein’s shape and charge (and hence its functional properties). Therefore some nonsynonymous substitutions are selectively neutral, or nearly so. A third possibility is that a nonsynonymous substitution alters a protein in a way that confers an advantage to the organism, and is therefore favored by natural selection.
LINK
The genetic code determines the amino acid that is encoded by each codon; see Figure 10.11
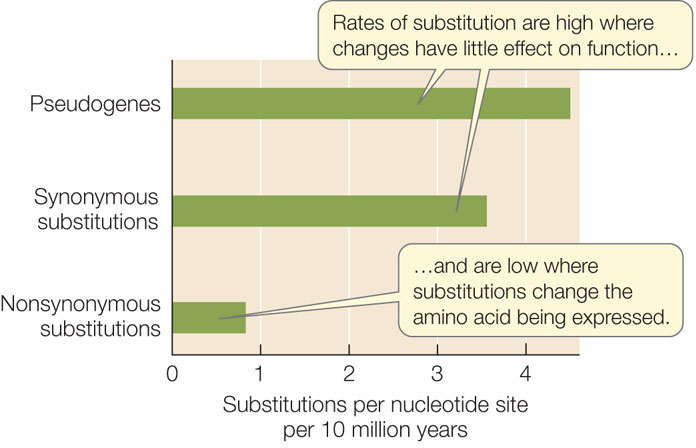
The rate of synonymous substitutions in most protein-coding genes is much higher than the rate of nonsynonymous substitutions. In other words, substitution rates are highest at nucleotide positions that do not change the amino acid being expressed (FIGURE 15.18). The rate of substitution is even higher in pseudogenes, which are copies of genes that are no longer functional.
Insertions, deletions, and rearrangements of DNA sequences are all mutations that may affect a larger portion of the gene or genome than do point mutations (see Concept 9.3). Insertions and deletions of nucleotides in a protein-coding sequence interrupt its reading frame, unless they occur in multiples of three nucleotides (the length of one codon). Rearrangements may merely change the order of whole genes along chromosomes, or they may rearrange functional domains among individual genes.
When biologists began to examine the details of genetic variation of populations, they soon discovered many gene variants that had little or no effect on function. This gave rise to new ideas about how these neutral variants arise and spread in populations.
Much of molecular evolution is neutral
Motoo Kimura proposed the neutral theory in 1968. He suggested that, at the molecular level, the majority of variants found in most populations are selectively neutral. That is, most gene variants confer neither an advantage nor a disadvantage on their bearers. Therefore these neutral variants must accumulate through genetic drift rather than through positive selection.
We saw in Concept 15.2 that genetic drift of existing gene variants tends to be greatest in small populations. However, the rate of fixation of new neutral mutations by genetic drift is independent of population size. To see why this is so, consider a population of size N and a neutral mutation rate of μ(mu) per gamete per generation at a particular locus. The number of new mutations would be, on average, μ × 2N, because 2N gene copies are available to mutate in a population of diploid organisms. The probability that a given mutation will be fixed by drift alone is its frequency, which equals 1/(2N) for a newly arisen mutation. We can multiply these two terms to get the rate of fixation of neutral mutations (m) in a given population of N individuals:
314
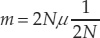
Therefore the rate of fixation of neutral mutations depends only on the neutral mutation rate μ and is independent of population size. Any given mutation is more likely to appear in a large population than in a small one, but any mutation that does appear is more likely to become fixed in a small population. These two influences of population size cancel each other out. Therefore the rate of fixation of neutral mutations is equal to the mutation rate (i.e., m = μ).
As long as the underlying mutation rate is constant, genes and proteins evolving in different populations should diverge from one another in neutral changes at a constant rate. The rate of evolution of particular genes and proteins is indeed often relatively constant over time, and therefore can be used as a “molecular clock” to calculate evolutionary divergence times between species (see Concept 16.3).
Neutral theory does not imply that most mutations have no effect on the individual organism, even though much of the genetic variation present in a population is the result of neutral evolution. Many mutations are never observed in populations because they are lethal or strongly detrimental, and the individuals that carry them are quickly removed from the population through natural selection. Similarly, because mutations that confer a selective advantage tend to be quickly fixed in populations, they also do not result in significant variation at the population level. Nonetheless, if we compare homologous proteins from different populations or species, some amino acid positions will remain constant under purifying selection, others will vary through neutral genetic drift, and still others will differ among species as a result of positive selection for change. How can these evolutionary processes be distinguished?
Positive and purifying selection can be detected in the genome
As we have just seen, substitutions in a protein-coding gene can be either synonymous or nonsynonymous, depending on whether they change the resulting amino acid sequence of the protein. The relative rates of synonymous and nonsynonymous substitutions are expected to differ in regions of genes that are evolving neutrally, or evolving under positive selection for change, or staying unchanged under purifying selection.
- If a given amino acid in a protein can be one of many alternatives (without changing the protein’s function), then an amino acid replacement is neutral with respect to the fitness of an organism. In this case, the rates of synonymous and nonsynonymous substitutions in the corresponding DNA sequences are expected to be very similar, so the ratio of the two rates should be close to 1.
- If a given amino acid position is under positive selection for change, the observed rate of nonsynonymous substitutions is expected to exceed the rate of synonymous substitutions in the corresponding DNA sequences.
- If a given amino acid position is under purifying selection, then the observed rate of synonymous substitutions is expected to be much higher than the rate of nonsynonymous substitutions in the corresponding DNA sequences.
The evolution of lysozyme illustrates how and why particular codons in a gene sequence might be under different modes of selection. The enzyme lysozyme is found in almost all animals. It is produced in the tears, saliva, and milk of mammals and in the albumen (whites) of bird eggs. Lysozyme digests the cell walls of bacteria, rupturing and killing them. As a result, it plays an important role as a first line of defense against invading bacteria. Most animals defend themselves against bacteria by digesting them, which is probably why most animals have lysozyme. Some animals also use lysozyme in the digestion of food.
Among mammals, a mode of digestion called foregut fermentation has evolved twice. In mammals with this mode of digestion, the foregut—consisting of part of the esophagus and/or stomach—has been converted into a chamber in which bacteria break down ingested plant matter by fermentation. Foregut fermenters can obtain nutrients from the otherwise indigestible cellulose that makes up a large proportion of plant tissue. Foregut fermentation evolved independently in ruminants (a group of hoofed mammals that includes cattle) and in certain leaf-eating monkeys, such as langurs. We know that these evolutionary events were independent because both langurs and ruminants have close relatives that are not foregut fermenters.
In both mammalian foregut-fermenting lineages, lysozyme has been modified to play a new, nondefensive role. The modified lysozyme enzyme ruptures some of the bacteria that live in the foregut, releasing nutrients metabolized by the bacteria, which the mammal then absorbs. How many changes in the lysozyme molecule were needed to allow it to perform this function amid the digestive enzymes and acidic conditions of the mammalian foregut? To answer this question, biologists compared the lysozyme-coding sequences in foregut fermenters with those in several of their nonfermenting relatives. They determined which amino acids differed and which were shared among the species (FIGURE 15.19A), as well as the rates of synonymous and nonsynonymous substitution in lysozyme genes across the evolutionary history of the sampled species.

For many of the amino acid positions of lysozyme, the rate of synonymous substitution in the corresponding gene sequence was much higher than the rate of nonsynonymous substitution. This observation indicates that many of the amino acids that make up lysozyme are evolving under purifying selection. In other words, there is selection against change in the lysozyme protein at these positions, and the encoded amino acids must therefore be critical for lysozyme function. At other positions, several different amino acids function equally well, and the corresponding codons have similar rates of synonymous and nonsynonymous substitution.
315
Genomes reveal both neutral and selective processes of evolution
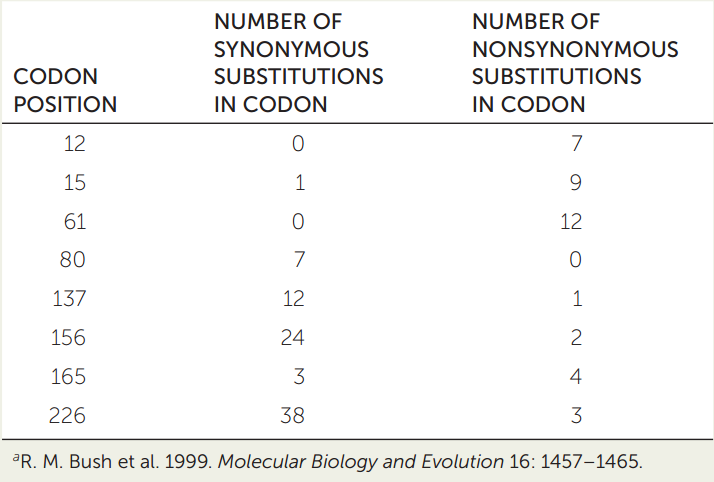
Analysis of synonymous and nonsynonymous substitutions in protein-coding genes can be used to detect neutral evolution, positive selection, and purifying selection. An investigator compared many gene sequences that encode the protein hemagglutinin (a surface protein of influenza virus) sampled over time, and collected the data at right.a Use the table to answer the following questions.
- Which codon positions are likely evolving under positive selection? Why?
- Which codon positions are likely evolving under purifying selection? Why?
(Hint: To calculate rates of each substitution type, you will need to consider the number of synonymous and nonsynonymous substitutions relative to the number of possible substitutions of each type. There are approximately three times as many possible nonsynonomous substitutions as there are synonymous substitutions.)
The most striking finding was that amino acid replacements in lysozyme happened at a much higher rate in the lineage leading to langurs than in any other primate lineage. The high rate of nonsynonymous substitution in the langur lysozyme gene shows that lysozyme went through a period of rapid change in adapting to the stomachs of langurs. Moreover, the lysozymes of langurs and cattle share five convergent amino acid replacements, all of which lie on the surface of the lysozyme molecule, well away from the enzyme’s active site. Several of these shared replacements are changes from arginine to lysine, which make the protein more resistant to degradation by the stomach enzyme pepsin. By understanding the functional significance of amino acid replacements, biologists can explain the observed changes in amino acid sequences in terms of changes in the functioning of the protein.
316
A large body of fossil, morphological, and molecular evidence shows that langurs and cattle do not share a recent common ancestor. However, langur and ruminant lysozymes share several amino acids that neither mammal shares with the lysozymes of its own closer relatives. The lysozymes of these two mammals have converged on some of the same amino acids despite their very different ancestry. The amino acids they share give these lysozymes the ability to lyse the bacteria that ferment plant material in the foregut.
The hoatzin, an unusual leaf-eating South American bird (FIGURE 15.19B) and the only known avian foregut fermenter, offers another remarkable example of the convergent evolution of lysozyme. Many birds have an enlarged esophageal chamber called a crop. The crop of the hoatzin contains lysozyme and bacteria and acts as a fermentation chamber. Many of the amino acid replacements that occurred in the adaptation of hoatzin lysozyme are identical to those that evolved in ruminants and langurs. Thus even though the hoatzin and foregut-fermenting mammals have not shared a common ancestor in hundreds of millions of years, similar adaptations have evolved in their lysozyme enzymes, enabling both groups to recover nutrients from fermenting bacteria.
Heterozygote advantage maintains polymorphic loci
In many cases, different alleles of a particular gene are advantageous under different environmental conditions. Most organisms, however, experience a wide diversity of environments. A night is dramatically different from the preceding day. A cold, cloudy day differs from a clear, hot one. Day length and temperature change seasonally. For many genes, a single allele is unlikely to perform well under all these conditions. In such situations, a heterozygous individual (with two different alleles) is likely to outperform individuals that are homozygous for either one of the alleles.
Colias butterflies of the Rocky Mountains live in environments where dawn temperatures often are too cold, and afternoon temperatures too hot, for the butterflies to fly. Populations of these butterflies are polymorphic for the gene that encodes phosphoglucose isomerase (PGI), an enzyme that influences how well an individual flies at different temperatures. Butterflies with certain PGI genotypes can fly better during the cold hours of early morning; those with other genotypes perform better during midday heat. The optimal body temperature for flight is 35°C–39°C, but some butterflies can fly with body temperatures as low as 29°C or as high as 40°C. Heat-tolerant genotypes are favored during spells of unusually hot weather; during spells of unusually cool weather, cold-tolerant genotypes are favored.
317
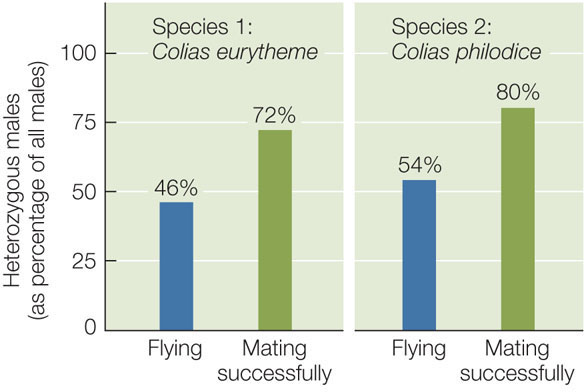
Heterozygous Colias butterflies can fly over a greater temperature range than homozygous individuals because they produce two different forms of PGI. This greater range of activity should give them an advantage in foraging and finding mates. A test of this prediction did find a mating advantage in heterozygous males, and further found that this mating advantage maintains the polymorphism in the population (FIGURE 15.20). The heterozygous condition can never become fixed in the population, however, because the offspring of two heterozygotes will always include both classes of homozygotes in addition to heterozygotes.
Investigation
HYPOTHESIS
Heterozygous male Colias will have proportionally greater mating success than homozygous males.
- For each of two Colias species, capture butterflies in the field. In the laboratory, determine their genotypes and allow them to mate.
- Determine the genotypes of the offspring, thus revealing paternity and mating success of the males.
For both species, the proportion of heterozygous males that mated successfully was higher than the proportion of all males seeking females (“flying”).
CONCLUSION
Heterozygous Colias males have a mating advantage over homozygous males.
ANALYZE THE DATA
Analyze these sampling data collected during the experiment (only one of several samples is shown for each species).

- Under the assumption that the proportions of each genotype (heterozygotes and homozygotes) of mating males are the same as the proportions seen among all viable males, calculate the number of mating males expected to be heterozygous and the number expected to be homozygous.
- Use a chi-square test (see Appendix B) to evaluate the significance of the difference in your expected numbers in (A) and the observed percentages of heterozygous mating males. The critical value (P = 0.05) of the chi-square distribution with one degree of freedom is 3.841. Are the observed and expected numbers of heterozygotes and homozygotes among mating males significantly different in these samples?
aW. B. Watt et al. 1985. Genetics 109: 157–175.
Genome size and organization also evolve
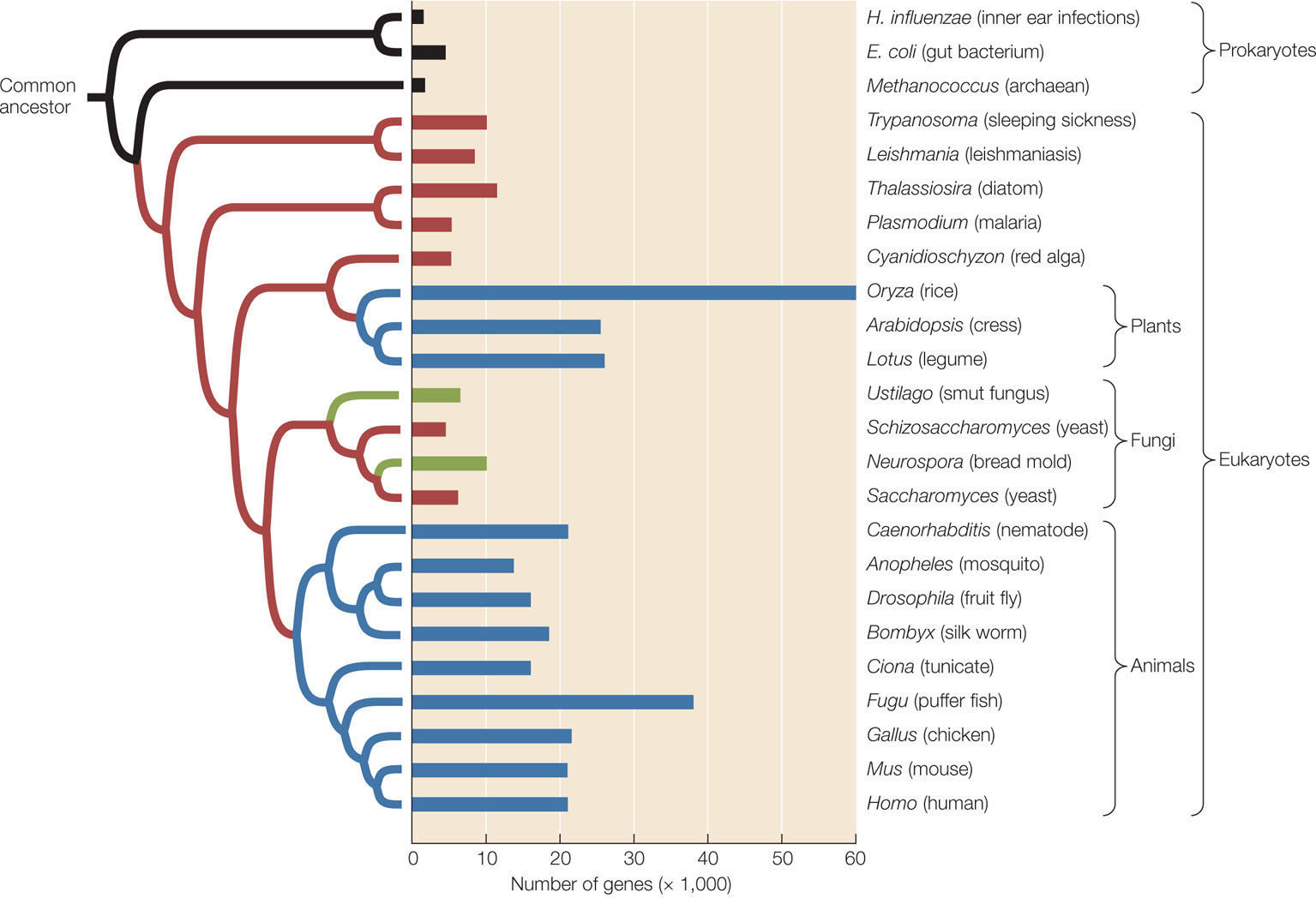
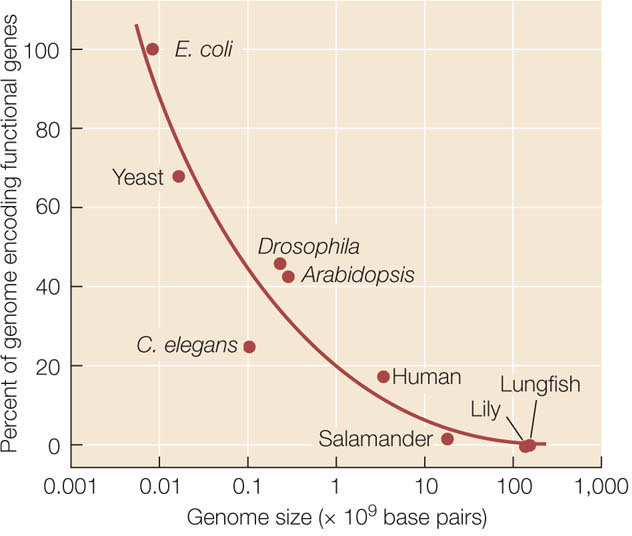
We know that genome size varies tremendously among organisms. Across broad taxonomic categories, there is some correlation between genome size and organismal complexity. The genome of the tiny bacterium Mycoplasma genitalium has only 470 genes. Rickettsia prowazekii, the bacterium that causes typhus, has 634 genes. Homo sapiens, by contrast, has about 21,000 protein-coding genes. FIGURE 15.21 shows the number of genes from a sample of organisms whose genomes have been fully sequenced, arranged by their evolutionary relationships. As this figure reveals, however, a larger genome does not always indicate greater complexity (compare rice with the other plants, for example). It is not surprising that more complex genetic instructions are needed for building and maintaining a large, multicellular organism than a small, single-celled bacterium. What is surprising is that some organisms, such as lungfishes, some salamanders, and lilies, have about 40 times as much DNA as humans do (FIGURE 15.22). Structurally, a lungfish or a lily is not 40 times more complex than a human. So why does genome size vary so much?
318
Differences in genome size are not so great if we take into account only the portion of DNA that actually encodes proteins. The organisms with the largest total amounts of nuclear DNA (some ferns and flowering plants) have 80,000 times as much DNA as do the bacteria with the smallest genomes, but no species has more than about 100 times as many protein-coding genes as a bacterium. Therefore much of the variation in genome size lies not in the number of functional genes, but in the amount of noncoding DNA (see Figure 15.22).
Why do the cells of most eukaryotic organisms have so much noncoding DNA? Does this noncoding DNA have a function? Although some of this DNA does not encode proteins, it can alter the expression of the genes surrounding it. The degree or timing of gene expression can vary dramatically depending on the gene’s position relative to noncoding sequences that regulate gene expression. Other regions of noncoding DNA consist of pseudogenes (regions that have evolved from functional genes, even though they have no function at present). Pseudogenes are often carried in the genome because the cost of doing so is very small. Occasionally, these pseudogenes become the raw material for the evolution of new genes with novel functions. Other noncoding sequences function in maintaining chromosomal structure. Still others consist of parasitic transposable elements that spread through populations because they reproduce faster than the host genome.
Another hypothesis is that the proportion of noncoding DNA is related primarily to population size. Noncoding sequences that are only slightly deleterious to the organism are likely to be purged by selection most efficiently in species with large population sizes. In species with small populations, the effects of genetic drift can overwhelm selection against noncoding sequences that have small deleterious consequences. Therefore selection against the accumulation of noncoding sequences is most effective in species with large populations, so such species (such as bacteria or yeasts) have relatively little noncoding DNA compared with species with small populations (see Figure 15.22).
CHECKpointCONCEPT15.5
- How can the ratio of synonymous to nonsynonymous substitutions be used to determine whether a particular gene is evolving neutrally, under positive selection, or under stabilizing selection?
- Why is the rate of fixation of neutral mutations independent of population size?
- Why do heterozygous individuals sometimes have an advantage over homozygous individuals?
- Why can a mutation that results in the replacement of one amino acid by another be a neutral event in some cases and in other cases be detrimental or beneficial? (Hint: Review the information about amino acids in Table 3.2 and the details of protein structure in Concept 3.2.)
- Postulate and contrast two hypotheses for the wide diversity of genome sizes among different organisms.
Most of our discussion so far has centered on changes in existing genes and phenotypes. Next we’ll consider how new genes with novel functions arise in populations in the first place.