41.3 Eukaryotic Genes Can Be Manipulated with Considerable Precision
Eukaryotic genes can be introduced into bacteria by using the techniques of recombinant DNA technology as heretofore discussed. The bacteria can then be used as factories to produce the desired gene product, usually a protein. Producing such a molecular factory will be our plan of attack for the estrogen receptor. First, however, we must generate DNA encoding the estrogen receptor.
Complementary DNA Prepared from mRNA Can Be Expressed in Host Cells
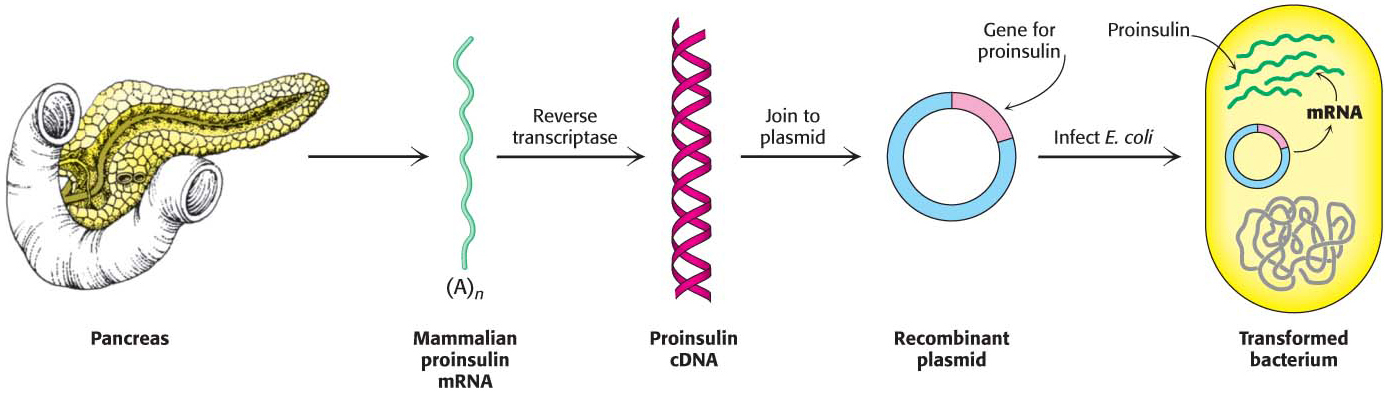
How can mammalian DNA be cloned and expressed by E. coli? Recall that most mammalian genes are mosaics of introns and exons. These interrupted genes cannot be expressed by bacteria, which lack the machinery to splice introns out of the primary transcript. However, this difficulty can be circumvented by introducing recombinant DNA that is complementary to mature mRNA, or cDNA, into the bacteria. For example, proinsulin, a precursor of insulin, is synthesized by bacteria harboring plasmids that contain DNA complementary to mRNA for proinsulin (Figure 41.6). Indeed, bacteria produce much of the insulin used today by millions of diabetics.
DID YOU KNOW?
Retroviruses contain an RNA genome but replicate through a DNA intermediate. The conversion of RNA information into DNA information is catalyzed by reverse transcriptase. Human immunodeficiency virus (HIV), the cause of AIDS, is a retrovirus.
QUICK QUIZ
Why are restriction enzymes such vital tools for recombinant DNA technology?
Because of their high degree of specificity, restriction enzymes allow precise cleavage of double-
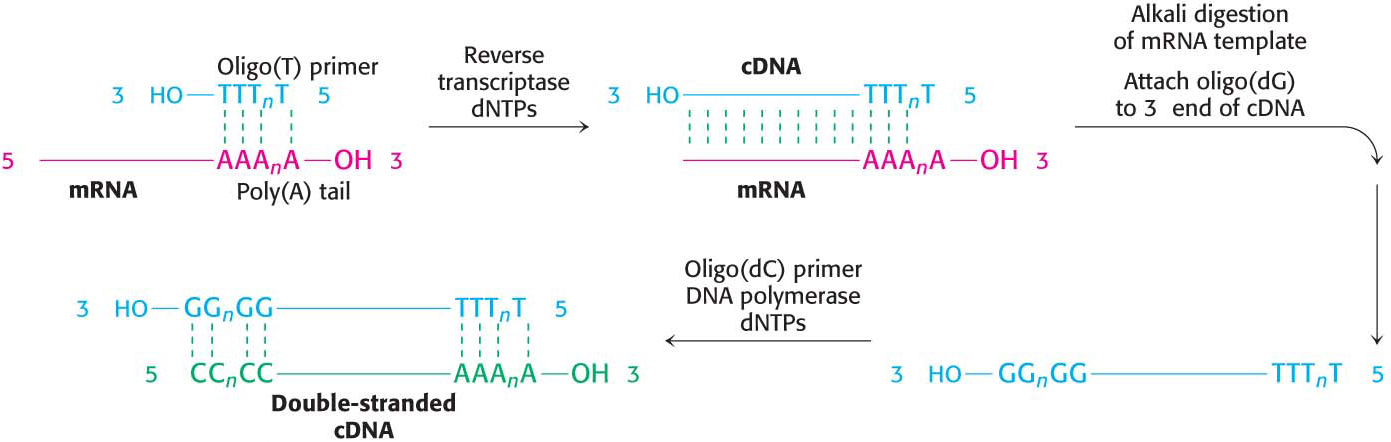
The key to forming complementary DNA is the enzyme reverse transcriptase, an RNA-
Estrogen-Receptor cDNA Can Be Identified by Screening a cDNA Library
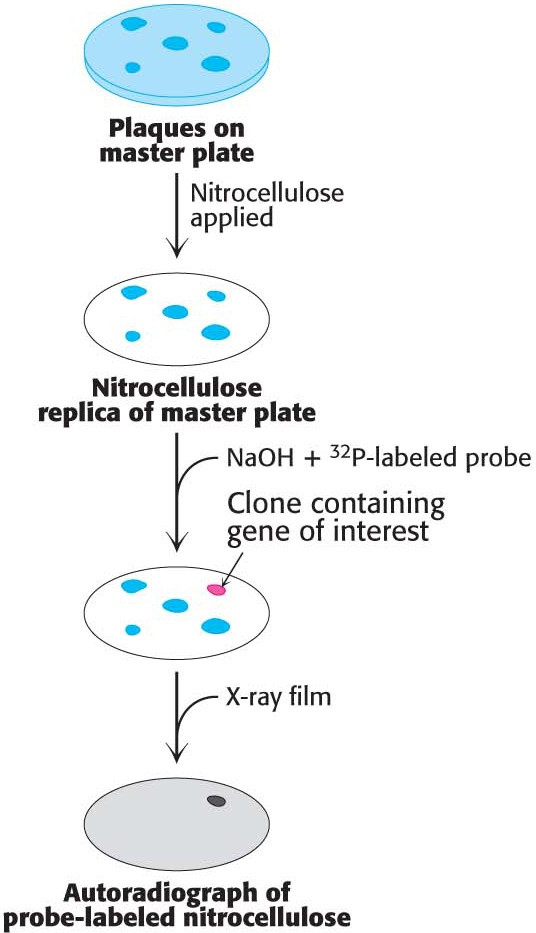
With these techniques at our disposal, let us return to our experiments with the estrogen receptor. We will use the probe that we synthesized earlier to screen a cDNA library generated from an estrogen-
A dilute suspension of the recombinant phages is first plated on a lawn of bacteria (Figure 41.8). Where each phage particle has landed and infected a bacterium, a plaque containing identical phages develops on the plate. A replica of this master plate is then made by applying a sheet of nitrocellulose. Infected bacteria and phage DNA released from lysed cells adhere to the sheet in a pattern of spots corresponding to the plaques. Intact bacteria on this sheet are lysed with NaOH, which also serves to denature the DNA so that it becomes accessible for hybridization with a 32P-
The vector containing the cDNA for the estrogen receptor can be isolated and transcribed. The resulting mRNA can be translated in vitro to produce receptor for experiments.
Complementary DNA Libraries Can Be Screened for Synthesized Protein
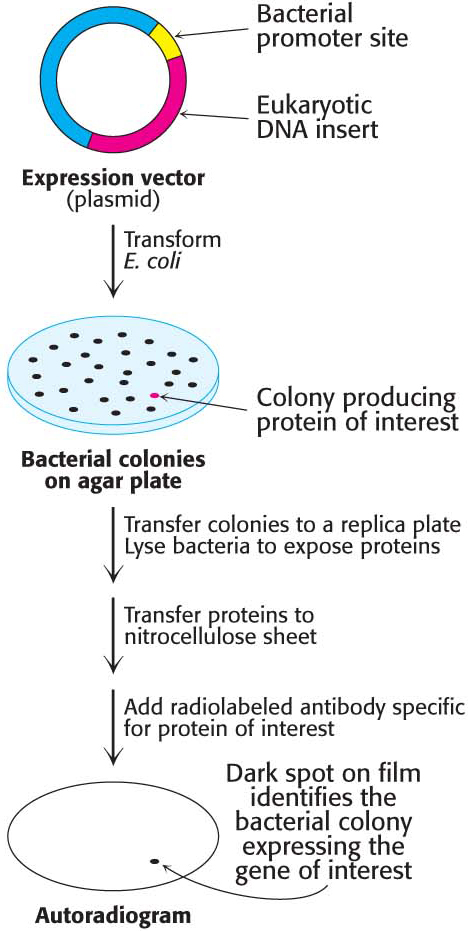
The vectors discussed so far simply carry the incorporated DNA and allow for the transcription of the inserted DNA. However, with the use of a specially prepared vector, bacteria that are actually expressing the estrogen-
Having a cDNA for the estrogen receptor enables us to perform a number of experiments to determine the biochemical properties of the protein. For instance, we learned earlier that the estrogen receptor is a transcription factor that functions by binding to the DNA of select genes. By using the cloned receptor, we can perform experiments to determine the DNA sequence to which the receptor binds most tightly. We could investigate whether the receptor reacts with other proteins when binding to DNA. Indeed, the knowledge to be gained is limited only by our imagination and experimental skill. However, having cDNA for the receptor tells us little about the gene that encodes the receptor itself. Does the gene contain introns? What regulatory sequences control its expression? To answer these questions and similar ones, we must isolate the gene that encodes the receptor. To do so, we return to a library, but this time to a genomic library.
Specific Genes Can Be Cloned from Digests of Genomic DNA
Let us see how we can clone a gene that is present just once in a haploid genome, such as the gene encoding the estrogen receptor. The approach is to prepare a large collection (library) of fragments of genomic DNA and then to identify those members of the collection that have the gene of interest.
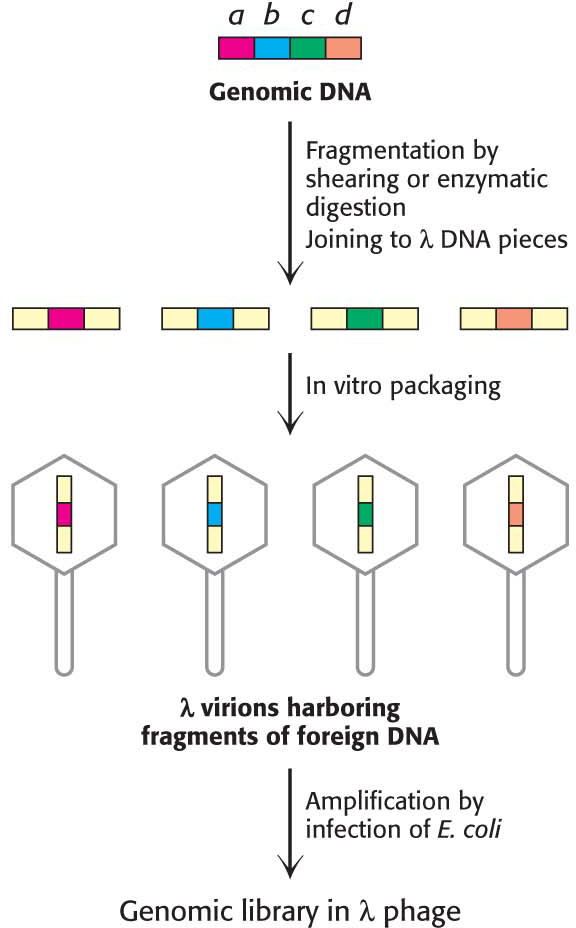
A sample containing many copies of total genomic DNA—
The gene of interest is unlikely to be found in one piece of DNA, because genes are usually larger than 15 kb, the size of the fragments used to make the genomic library. Consequently, several clones from the genomic library will harbor different parts of the gene for the estrogen receptor. These clones must be isolated and sequenced to determine the sequence of the entire gene.
DNA Can Be Sequenced by the Controlled Termination of Replication
The analysis of DNA structure and its role in gene expression also have been markedly facilitated by the development of powerful techniques for the sequencing of DNA molecules. The key to DNA sequencing is the generation of DNA fragments whose length is determined by the last base in the sequence. Collections of such fragments can be generated through the controlled termination of replication (Sanger dideoxy method), a method developed by Frederick Sanger and his coworkers. The same procedure is performed on four reaction mixtures at the same time. In all these mixtures, a DNA polymerase is used to make the complement of a short sequence within a single-
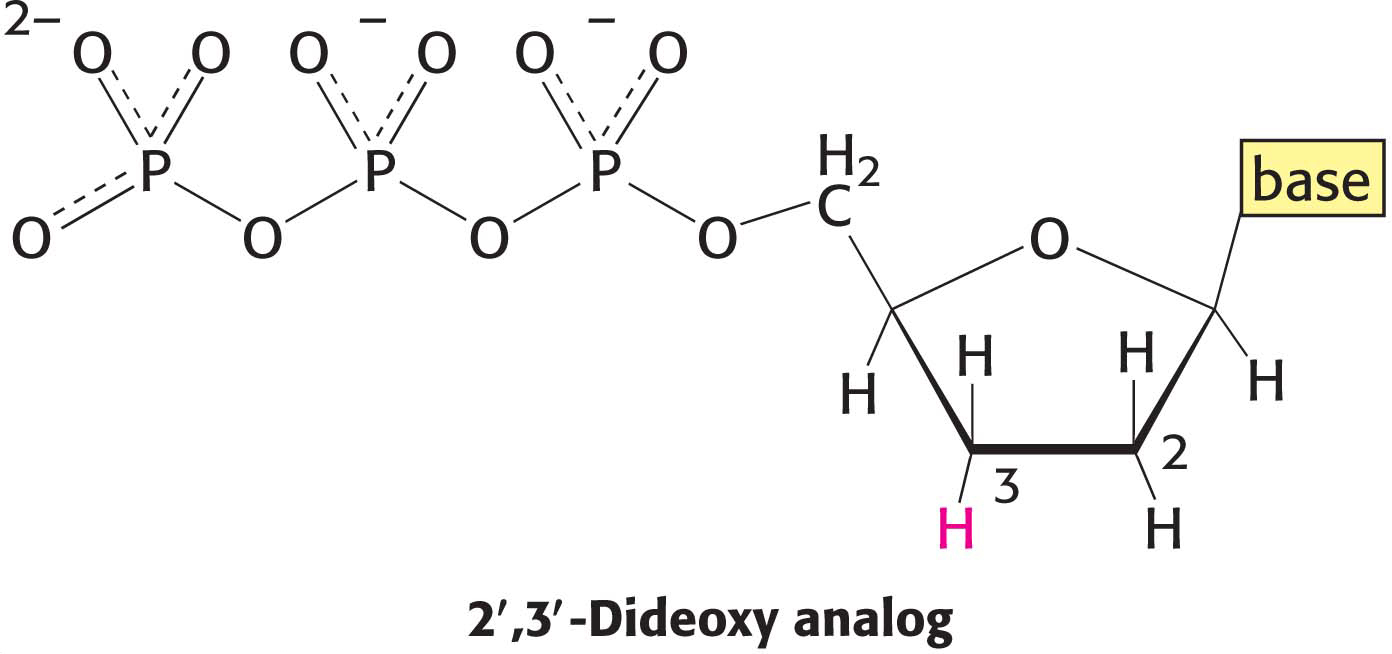
The incorporation of this analog blocks further growth of the new strand because the dideoxy analog lacks the 3′-hydroxyl terminus needed to form the next phosphodiester linkage. The concentration of the analog is low enough that strand termination will take place only occasionally. The polymerase will insert the correct nucleotide sometimes and the dideoxy analog other times, stopping the reaction. For instance, if the dideoxy analog of dATP (ddATP) is present, fragments of various lengths are produced, but all will be terminated by ddATP (Figure 41.11). Importantly, ddATP will be inserted only where a T was located in the DNA being sequenced. Thus, the fragments of different length will correspond to the positions of T. Four such sets of strand-
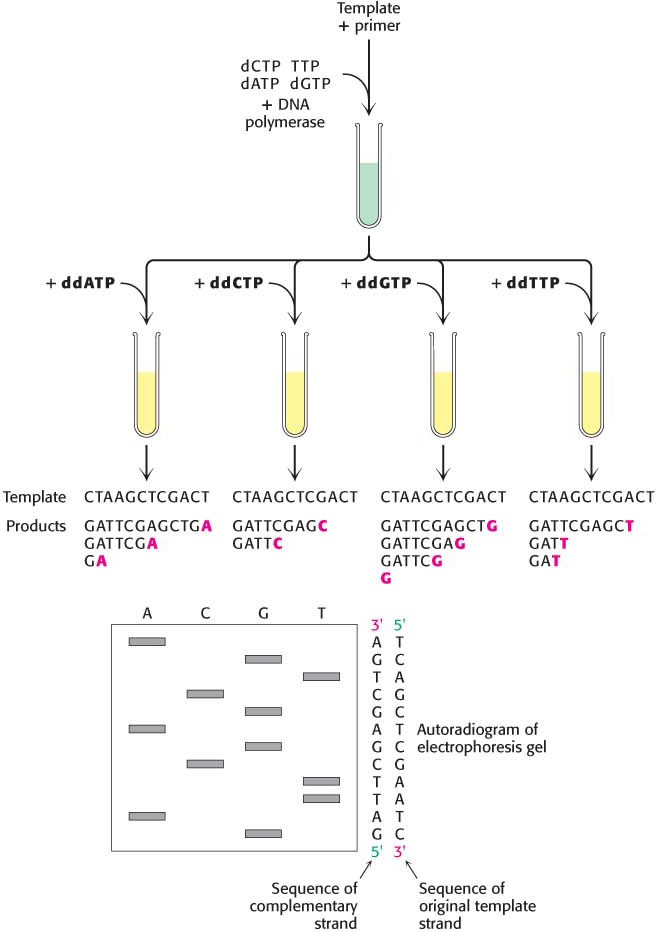
Fluorescence detection is a highly effective alternative to autoradiography. A fluorescent tag is incorporated into each dideoxy analog—
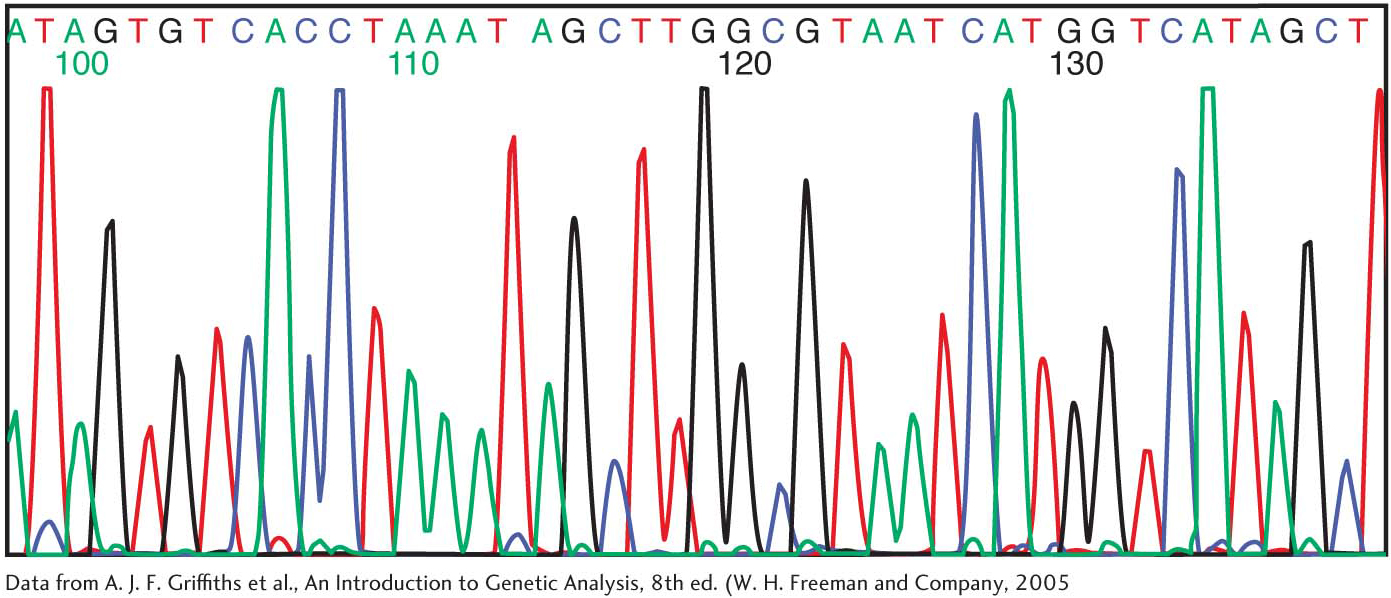
Applying such sequencing tools to the investigation of the gene for the estrogen receptor reveals that the gene is more than 140 kb in length and contains eight exons. In addition to TATA and CAAT boxes, the upstream region of the gene contains a P1 promoter that is activated by the transcription factor AP2γ. Interestingly, certain breast cancers depend on the presence of the estrogen receptor for malignant growth, and AP2γ may play a critical role in the regulation of the gene for the estrogen receptor in cancer cells.

 CLINICAL AND BIOLOGICAL INSIGHT
CLINICAL AND BIOLOGICAL INSIGHTNext-Generation Sequencing Methods Enable the Rapid Determination of a Complete Genome Sequence
Since the introduction of the Sanger dideoxy method in the mid-
Next-
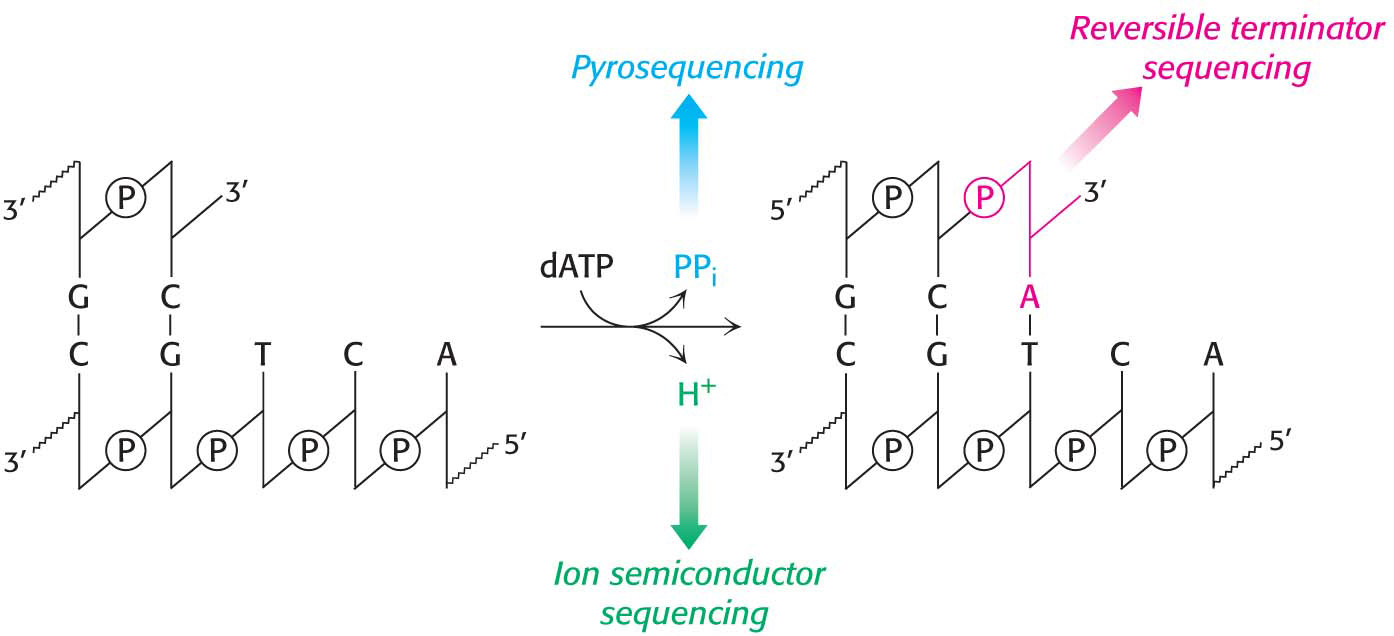
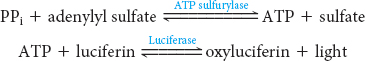
In pyrosequencing, nucleotides are added to the template DNA, one at a time in a defined order. One of the nucleotides will be incorporated into the growing strand, releasing a pyrophosphate that is detected by coupling the formation of pyrophosphate with the production of light by the sequential action of the enzymes ATP sulfurylase and luciferase:
The protocol for ion semiconductor sequencing is similar to pyrosequencing except that nucleotide incorporation is detected by sensitively measuring the very small changes in pH of the reaction mixture due to the release of proton upon nucleotide incorporation.
Regardless of the sequencing method, the technology exists to quantify the signal produced by millions of DNA fragment templates simultaneously. However, for many approaches, as few as 50 bases are read per fragment. Hence, significant computing power is required to both store the massive amounts of sequence data and perform the necessary alignments required to assemble a complete sequence. Next-
Selected DNA Sequences Can Be Greatly Amplified by the Polymerase Chain Reaction
Let us summarize our research accomplishments thus far. We have purified the estrogen receptor by using the monoclonal antibody that we generated (Chapter 5). We have synthesized a DNA probe that allowed us to isolate the cDNA of the receptor as well as the gene for the receptor. Finally, we have deduced the DNA sequence of the gene. Although there are many possible experiments to perform on the basis of what we have accomplished so far, let us start a new research project that will introduce us to one of the most powerful techniques in experimental biochemistry. Our experimental system thus far has been with the rat uterus. We can ask whether the receptor gene is transcribed in other tissues, such as the brain and the liver.
We could, in fact, screen cDNA libraries from these tissues, searching for a clone that contains the receptor cDNA as heretofore described. However, we will use a much more rapid means of detection. We will study cDNA prepared from brain and liver tissues as well as other tissues and will determine, with the use of the polymerase chain reaction (PCR), whether cDNA (and, by implication, the mRNA) for the receptor is present.
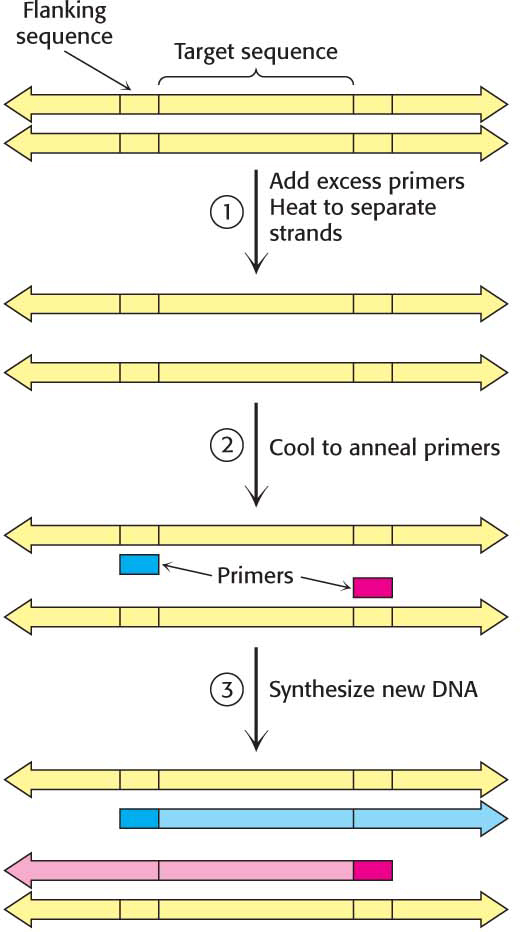
Consider a DNA duplex consisting of a target sequence surrounded by nontarget DNA. In our example, the target would be the putative receptor cDNA in the brain, the liver, or muscle. If the target DNA is present, we can detect it if we first amplify the amount of DNA present. Millions of copies of the target sequences can be readily obtained by PCR if the flanking sequences of the target are known, and we know what the flanking sequences are because we have the DNA sequence of the receptor. PCR is carried out by adding the following components to a solution containing the target sequence: (1) a pair of primers that hybridize with the flanking sequences of the target, (2) all four deoxyribonucleoside triphosphates (dNTPs), and (3) a heat-
Strand Separation. The two strands of the parent DNA molecule are separated by heating the solution to 95°C for 15 s.
Page 755Hybridization of Primers. The solution is then abruptly cooled to 54°C to allow each primer to hybridize to a DNA strand. One primer hybridizes to the 3′ end of the target on one strand, and the other primer hybridizes to the 3′ end on the complementary target strand. Parent DNA duplexes do not form, because the primers are present in large excess. Primers are typically from 20 to 30 nucleotides long.
DNA Synthesis. The solution is then heated to 72°C, the optimal temperature for Taq DNA polymerase. This heat-
stable polymerase comes from Thermus aquaticus, a thermophilic bacterium that lives in hot springs. The polymerase elongates both primers in the direction of the target sequence because DNA synthesis is in the 5′-to- 3′ direction. DNA synthesis takes place on both strands but extends beyond the target sequence.
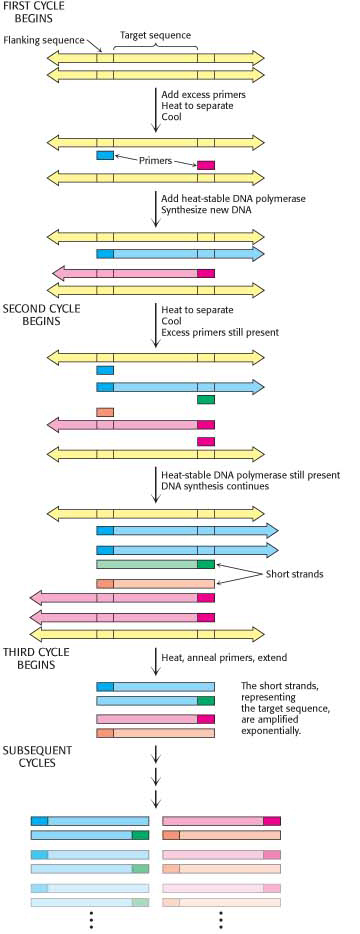
These three steps—
Several features of this remarkable method for amplifying DNA are noteworthy. First, the sequence of the target need not be known. All that is required is knowledge of the flanking sequences. Second, the target can be much larger than the primers. Targets larger than 10 kb have been amplified by PCR. Third, primers do not have to be perfectly matched to flanking sequences to amplify targets. With the use of primers derived from a gene of known sequence, it is possible to search for variations on the theme. In this way, families of genes are being discovered with the use of PCR. Fourth, PCR is highly specific because of the stringency of hybridization at relatively high temperature. Stringency is the required closeness of the match between primer and target, which can be controlled by temperature and salt. At high temperatures, the only DNA that is amplified is that situated between primers. A gene constituting less than a millionth of the total DNA of a higher organism is accessible by PCR. Fifth, PCR is exquisitely sensitive. A single DNA molecule can be amplified and subsequently visualized in gel electrophoresis. Indeed, the amplified DNA can be isolated from the gel and inserted into a vector and cloned if so desired.
PCR examination for the presence of the estrogen receptor in cDNA libraries from various tissues reveals that significant amounts of receptor mRNA are present in pituitary, bone, liver, and muscle cells, as well as in the reproductive tissues, including ovary, mammary gland, and uterus. Further studies using the same techniques show that the estrogen receptor is found in all vertebrates.
CLINICAL AND BIOLOGICAL INSIGHTPCR Is a Powerful Technique in Medical Diagnostics, Forensics, and Studies of Molecular Evolution
PCR can provide valuable diagnostic information in medicine. Bacteria and viruses can be readily detected with the use of specific primers. For example, PCR can reveal the presence of human immunodeficiency virus in people who have not mounted an immune response to this pathogen and would therefore be missed with an antibody assay. Finding Mycobacterium tuberculosis bacilli, the cause of tuberculosis, in tissue specimens is slow and laborious. With PCR, as few as 10 tubercle bacilli per million human cells can be readily detected. PCR is a promising method for the early detection of certain cancers. This technique can identify mutations of certain growth-
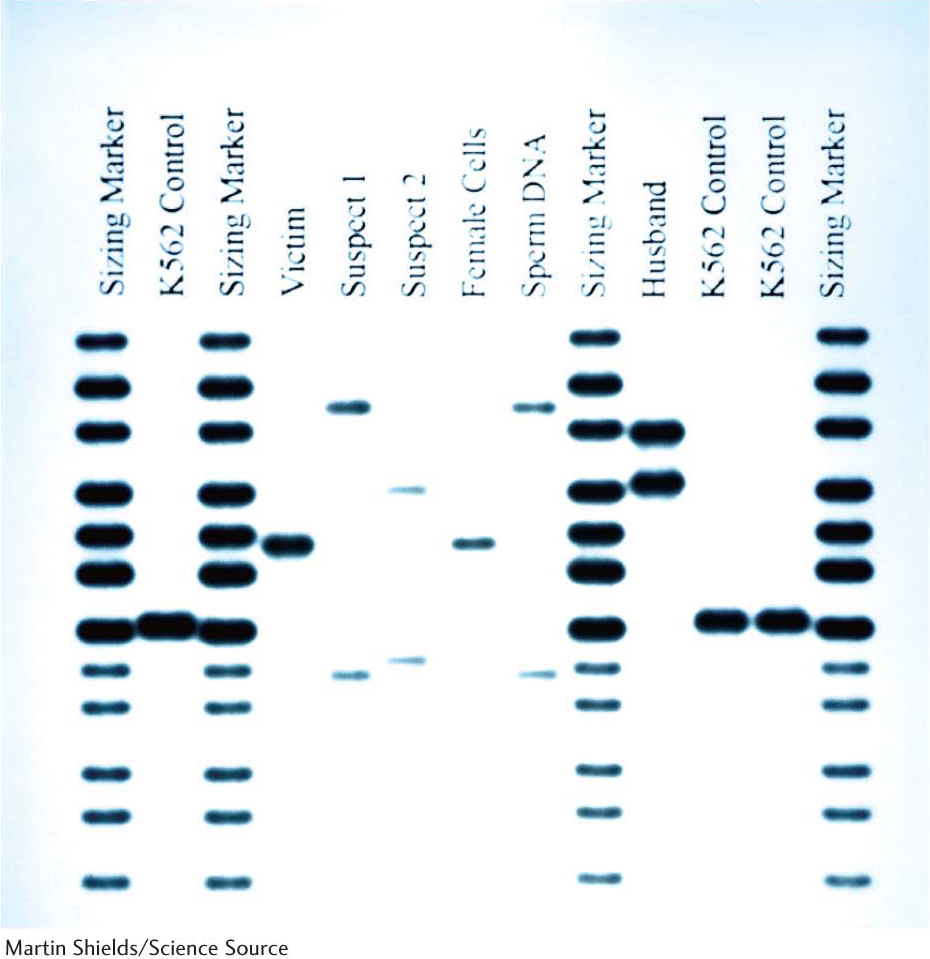
PCR is also having an effect in forensics and legal medicine. An individual DNA profile is highly distinctive because many genetic loci are highly variable within a population. For example, variations at specific loci determine a person’s HLA type (human-
Gene-Expression Levels Can Be Comprehensively Examined
Let us look now at a final technique, one that enables us to see how environmental signals, such as the presence of hormones, or pathological conditions, such as cancer, alter the expression of an array of genes in a tissue. Most genes are present in the same quantity in every cell—
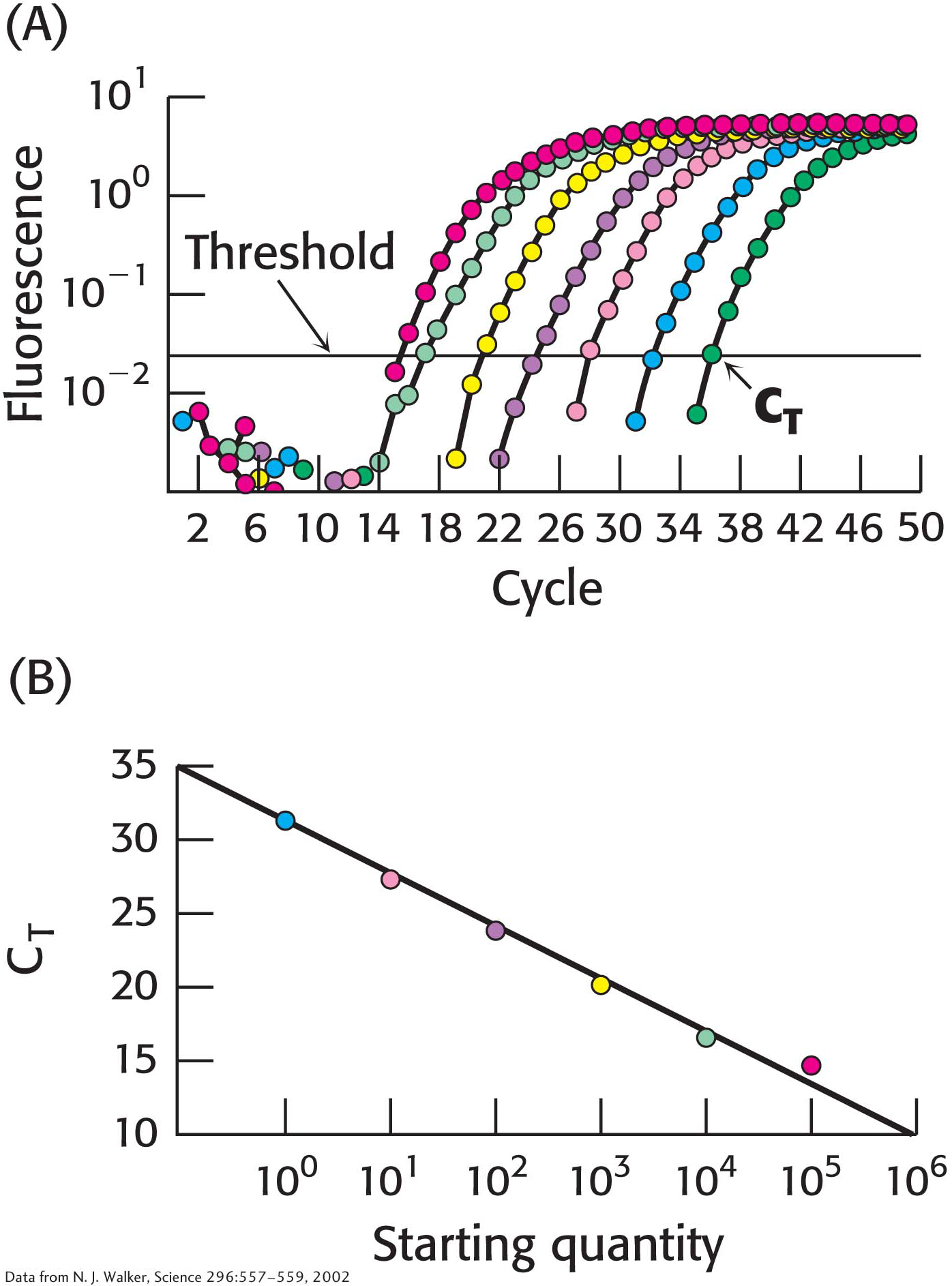
The quantity of individual mRNA transcripts can be determined by quantitative PCR (qPCR), or real-
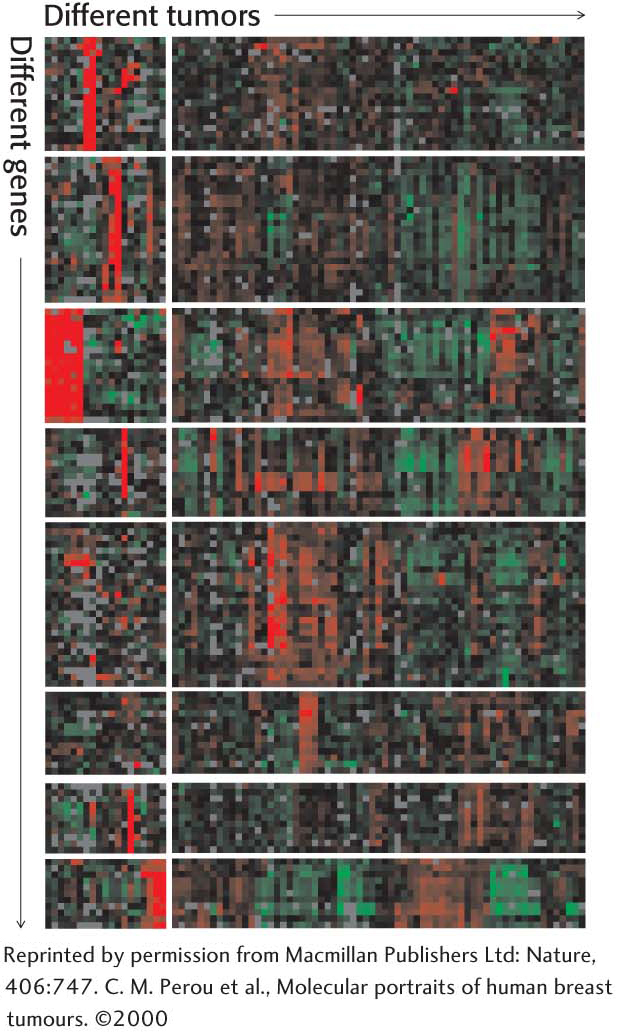
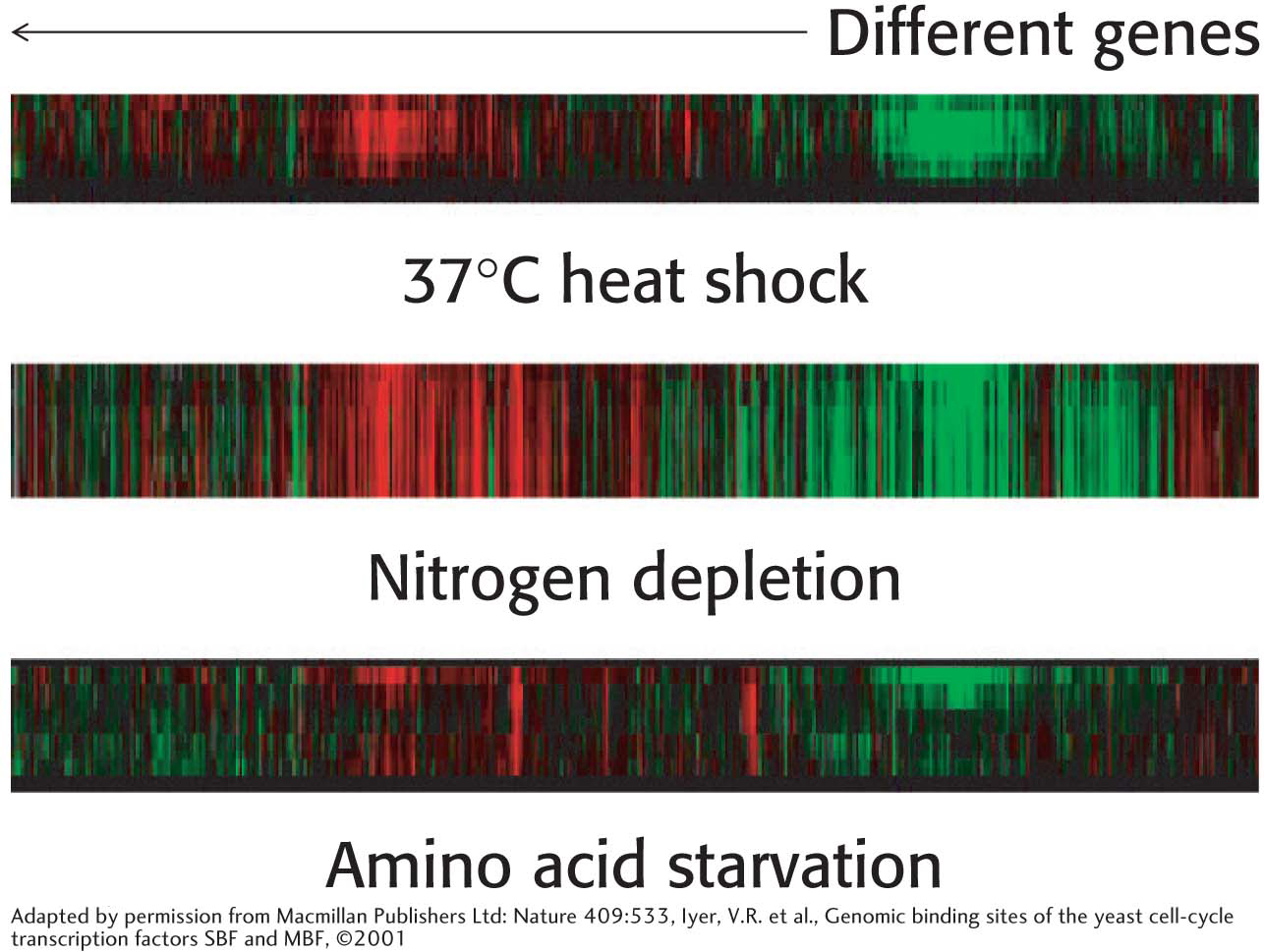
Although qPCR is a powerful technique for quantitation of a small number of transcripts in any given experiment, we can now use our knowledge of complete genome sequences to investigate an entire transcriptome, the pattern and level of expression of all genes in a particular cell or tissue. One of the most powerful methods developed to date for this purpose is based on hybridization. Oligonucleotides or cDNAs are affixed to a solid support such as a microscope slide, creating a DNA microarray, or gene chip. Fluorescently labeled cDNA is then hybridized to the chip to reveal the expression level for each gene, identifiable by its known location on the chip. The intensity of the fluorescent spot on the chip reveals the extent of the transcription of a particular gene. Figure 41.18 shows the pattern of genes that are induced or repressed in various breast-