
5.2 The Purification of a Protein Is the First Step in Understanding Its Function
✓ 4 Explain how proteins can be purified.
To understand a protein—
Proteins Can Be Purified on the Basis of Differences in Their Chemical Properties
Protein purification requires much ingenuity and patience, but, before we can even undertake the task, we need a test that identifies the protein that we are interested in. We will use this test after each stage of purification to see if the purification is working. Such a test is called an assay, and it is based on some unique identifying property of the protein. For enzymes, which are protein catalysts (Chapter 6), the assay is usually based on the reaction catalyzed by the enzyme in the cell. For instance, the enzyme lactate dehydrogenase, an important enzyme in glucose metabolism, carries out the following reaction:
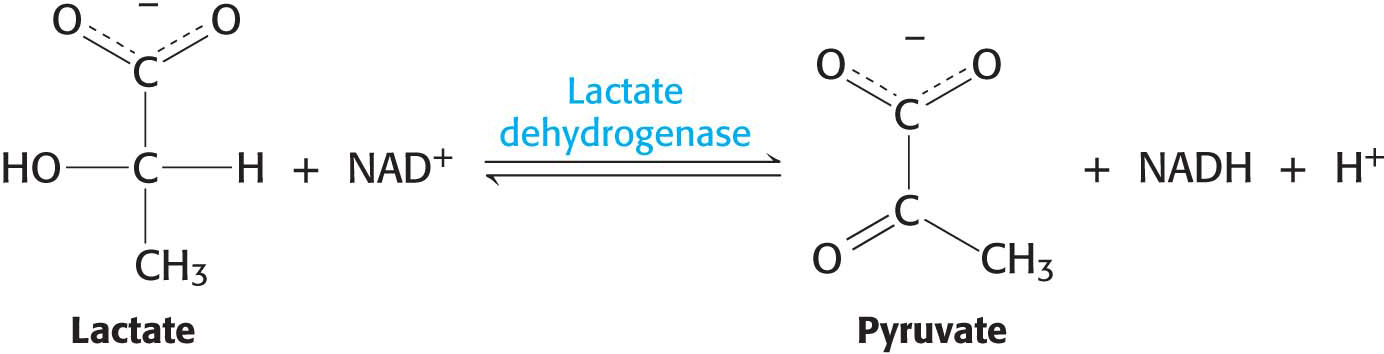
The product, reduced nicotinamide adenine dinucleotide (NADH), in contrast with the other reaction components, absorbs light at 340 nm. Consequently, we can follow the progress of the reaction by measuring the light absorbance at 340 nm in unit time—
QUICK QUIZ 1
Why is an assay required for protein purification?
An assay, which should be based on some unique biochemical property of the protein that is being purified, allows the detection of the protein of interest.
To be certain that our purification scheme is working, we need one additional piece of information—
Proteins Must Be Removed from the Cell to Be Purified
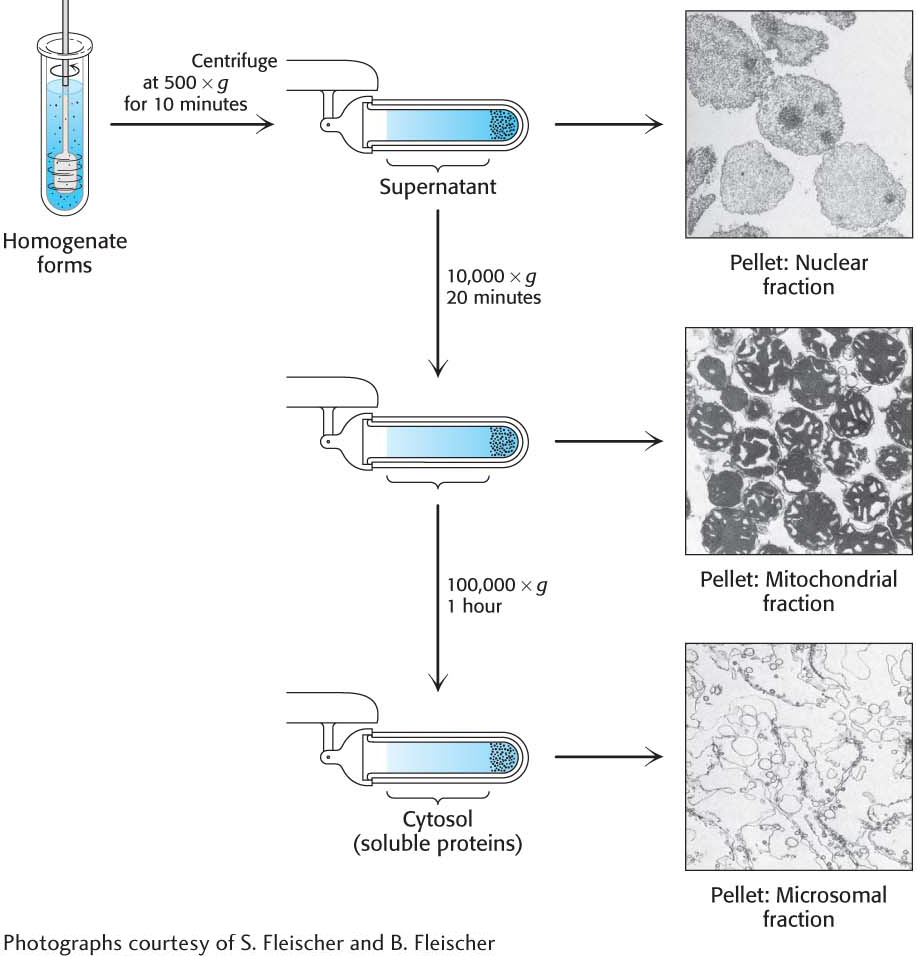
Having found an assay, we must now break open the cells, releasing the cellular contents, so that we can gain access to our protein. The disruption of the cell membranes yields a homogenate, a mixture of all of the components of the cell but no intact cells. This mixture is centrifuged at low centrifugal force, yielding a pellet of heavy material at the bottom of the centrifuge tube and a lighter solution above, called the supernatant. The pellets are enriched in a particular organelle (Figure 5.1). The pellet and supernatant are referred to as fractions because we are fractionating the homogenate. The supernatant is centrifuged again at a greater force to yield yet another pellet and supernatant. This procedure, called differential centrifugation, yields several fractions of decreasing density, each still containing hundreds of different proteins, which are assayed for the activity being purified. Usually, one fraction will have more enzyme activity than any other fraction, and it then serves as the source of material to which more-
Proteins Can Be Purified According to Solubility, Size, Charge, and Binding Affinity
Proteins are purified on the basis of differences in solubility, size, charge, and specific binding affinity. Usually, protein mixtures are subjected to a series of separations, each based on a different property.

Salting outMost proteins require some salt to dissolve, a process called salting in. However, most proteins precipitate out of solution at high salt concentrations, an effect called salting out (Figure 5.2). Salting out is due to competition between the salt ions and the protein for water to keep the protein in solution (water of solvation). The salt concentration at which a protein precipitates differs from one protein to another. Hence, salting out can be used to fractionate a mixture of proteins. Unfortunately, many proteins lose their activity in the presence of such high concentrations of salt. However, the salt can be removed by the process of dialysis. The protein–

Separation by sizeMolecular exclusion chromatography, also called gel-

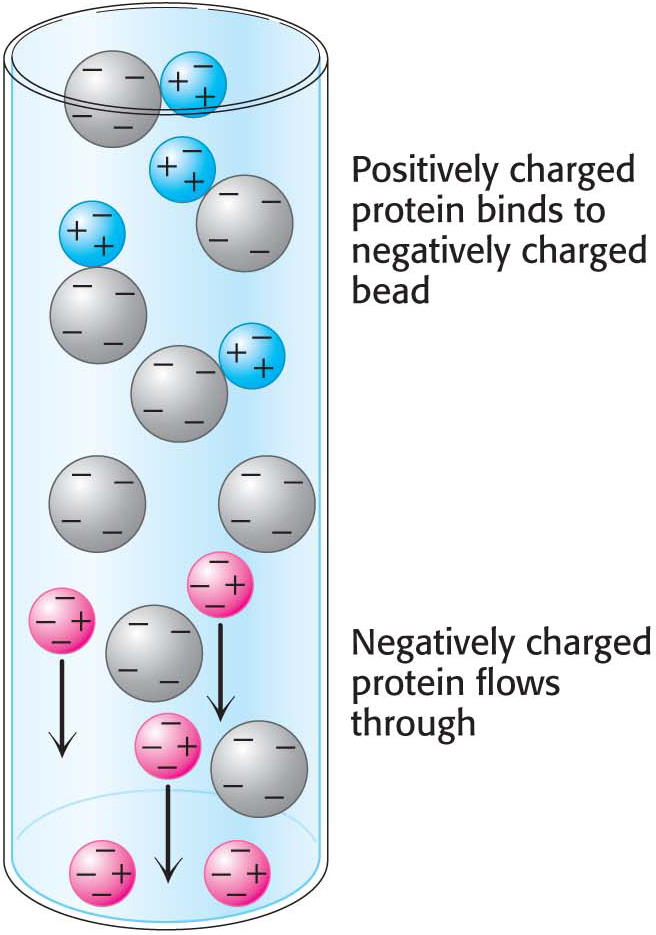
Ion-exchange chromatographyProteins can be separated on the basis of their net charge by ion-
Affinity chromatographyAffinity chromatography is another powerful means of purifying proteins. This technique takes advantage of the fact that some proteins have a high affinity for specific chemical groups or specific molecules. For example, the plant protein concanavalin A, which binds to glucose reversibly, can be purified by passing a crude extract through a column of beads containing covalently attached glucose residues. Concanavalin A binds to such beads, whereas most other proteins do not. The bound concanavalin A can then be released from the column by adding a concentrated solution of glucose. The glucose in solution displaces the column-


High-pressure liquid chromatographyThe ability of column techniques to separate individual proteins, called the resolving power, can be improved substantially through the use of a technique called high-
Proteins Can Be Separated by Gel Electrophoresis and Displayed
How can we tell whether a purification scheme is effective? One way is to demonstrate that the specific activity rises with each purification step. Another is to visualize the number of proteins present at each step. The technique of gel electrophoresis makes the latter method possible.
A molecule with a net charge will move in an electric field, a phenomenon termed electrophoresis. The distance and speed that a protein moves in electrophoresis depends on the electric-



Proteins can be separated largely on the basis of mass by electrophoresis in a polyacrylamide gel in the presence of the detergent sodium dodecyl sulfate (SDS), a technique called SDS–
Isoelectric focusingProteins can also be separated electrophoretically on the basis of their relative contents of acidic and basic residues. The isoelectric point (pI) of a protein is the pH at which its net charge is zero. At this pH, the protein will not migrate in an electric field. If a mixture of proteins is subjected to electrophoresis in a pH gradient in a gel in the absence of SDS, each protein will move until it reaches a position in the gel at which the pH is equal to the pI of the protein. This method of separating proteins is called isoelectric focusing. Proteins differing by one net charge can be separated (Figure 5.10).

Two-dimensional electrophoresisIsoelectric focusing can be combined with SDS–
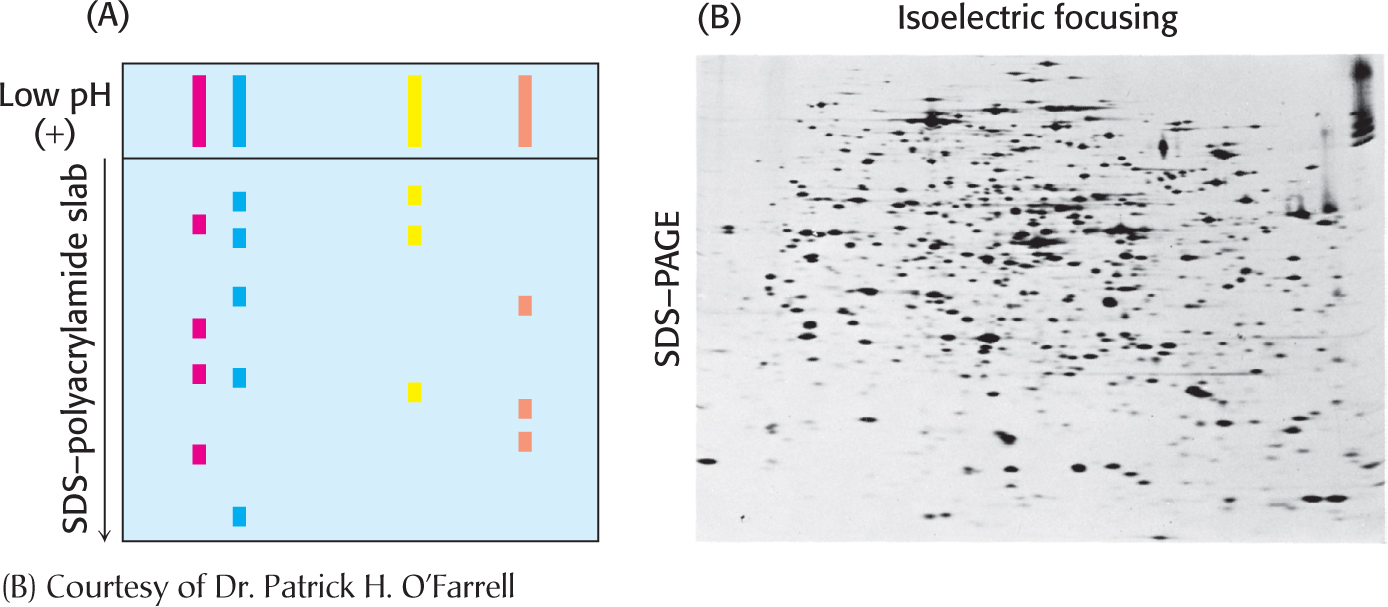
Proteins isolated from cells under different physiological conditions can be subjected to two-

A Purification Scheme Can Be Quantitatively EvaluatedSome combination of purification techniques will usually yield a pure protein. To determine the success of a protein-
Total Protein. The quantity of protein present in a fraction is obtained by determining the protein concentration of a portion of the fraction and multiplying by the fraction’s total volume.
Total Activity. The enzyme activity for the fraction is obtained by measuring the enzyme activity in the volume of fraction used in the assay and multiplying by the fraction’s total volume.
Specific Activity. This parameter, obtained by dividing total activity by total protein, enables us to measure the degree of purification by comparing specific activities after each purification step. Recall that the goal of a purification scheme is to maximize specific activity.
Page 78Yield. This parameter is a measure of the total activity retained after each purification step as a percentage of the activity in the crude extract. The amount of activity in the initial extract is taken to be 100%.
Purification Level. This parameter is a measure of the increase in purity and is obtained by dividing the specific activity, calculated after each purification step, by the specific activity of the initial extract.


As we see in Table 5.1, several purification steps can lead to several thousandfold purification. Inevitably, in each purification step, some of the protein of interest is lost, and so our overall yield is 35%. A good purification scheme takes into account purification levels as well as yield.
QUICK QUIZ 2
What physical differences among proteins allow for their purification?
Differences in size, solubility, charge, and the specific binding of certain molecules.
The SDS-