
9.6 Mutations in Genes Encoding Hemoglobin Subunits Can Result in Disease
One of the first diseases understood at the molecular level was the blood disease sickle-

 CLINICAL INSIGHT
CLINICAL INSIGHTSickle-Cell Anemia Is a Disease Caused by a Mutation in Hemoglobin
In 1904, James Herrick, a Chicago physician, examined a 20-
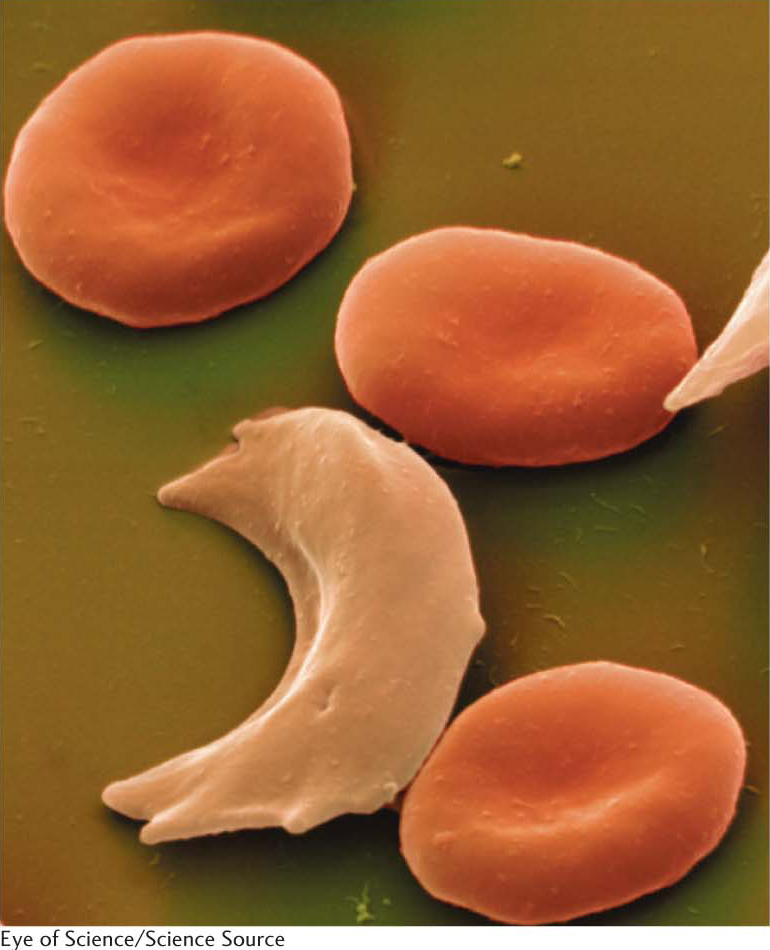
The patient’s blood smear contained unusual red cells, which Herrick described as sickle shaped (Figure 9.18). Other cases of this disease, called sickle-
Sickle-
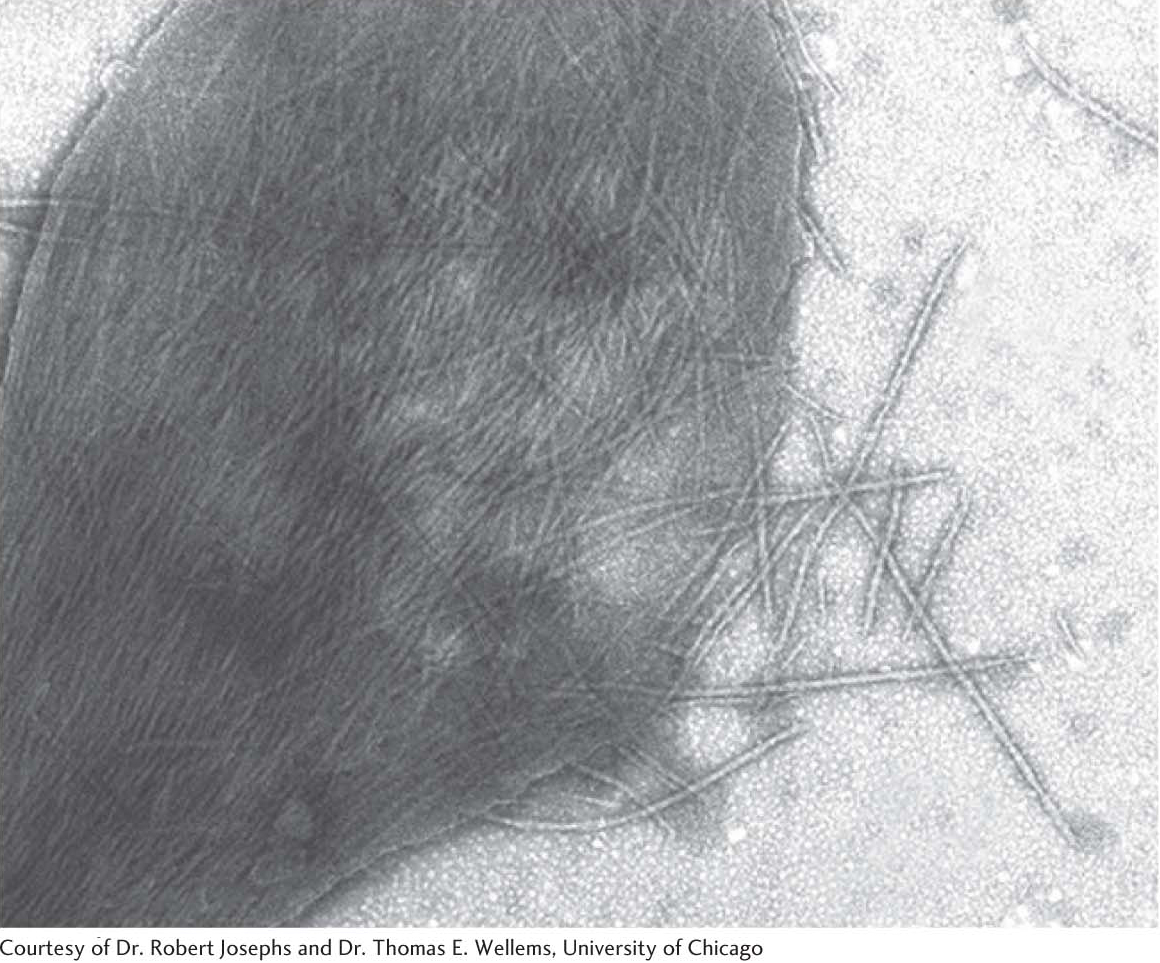
Examination of the contents of sickled red blood cells reveals that hemoglobin molecules have bound together to form large fibrous aggregates that extend across the cell, deforming the red cells and giving them their sickle shape (Figure 9.19). Sickle-
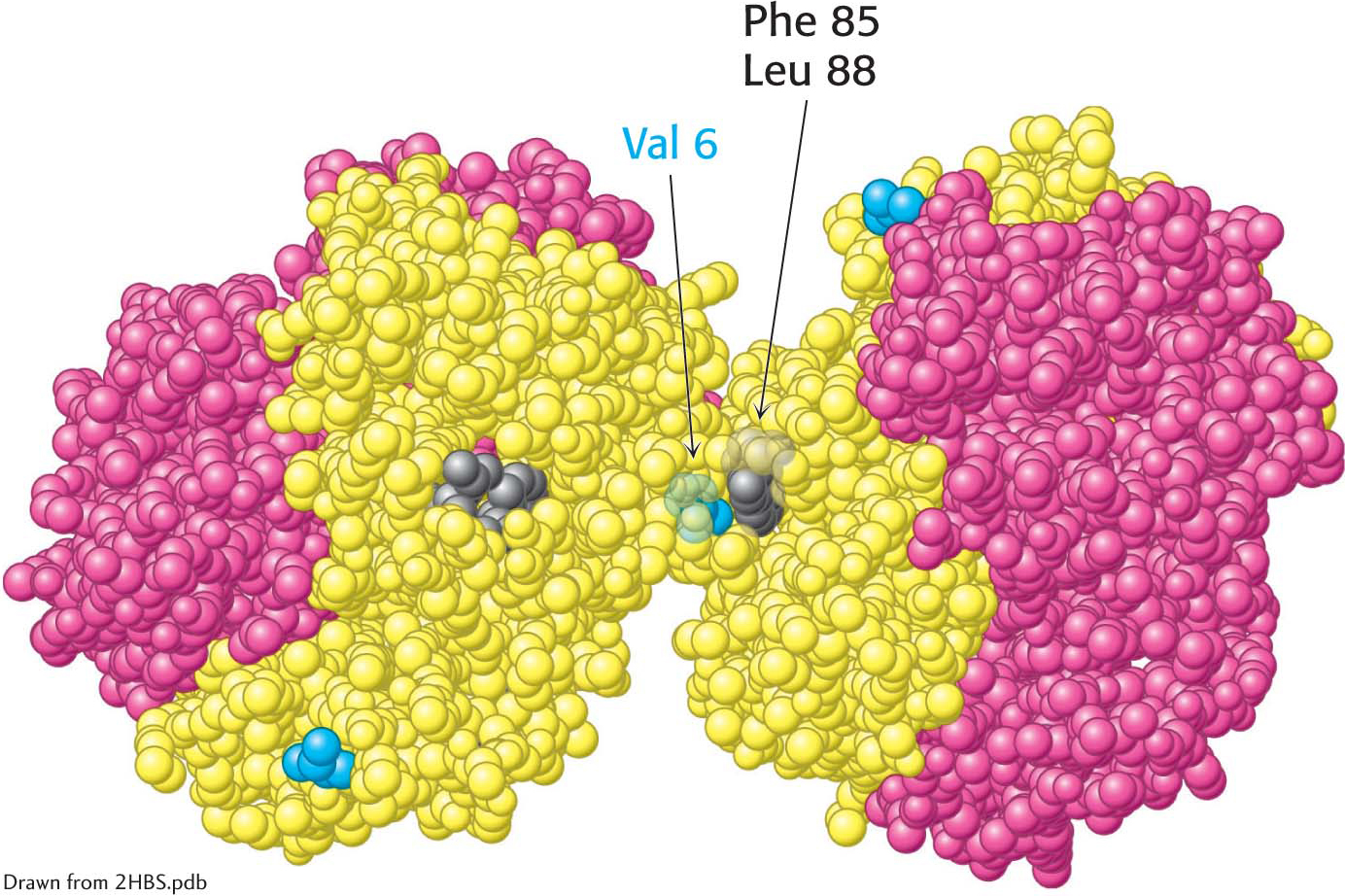
 Deoxygenated hemoglobin S. The interaction between valine 6 (blue) on a β chain of one hemoglobin molecule and a hydrophobic patch formed by phenylalanine 85 and leucine 88 (gray) on a β chain of another deoxygenated hemoglobin molecule leads to hemoglobin aggregation. The exposed valine 6 residues of other β chains participate in other such interactions in HbS fibers.
Deoxygenated hemoglobin S. The interaction between valine 6 (blue) on a β chain of one hemoglobin molecule and a hydrophobic patch formed by phenylalanine 85 and leucine 88 (gray) on a β chain of another deoxygenated hemoglobin molecule leads to hemoglobin aggregation. The exposed valine 6 residues of other β chains participate in other such interactions in HbS fibers.
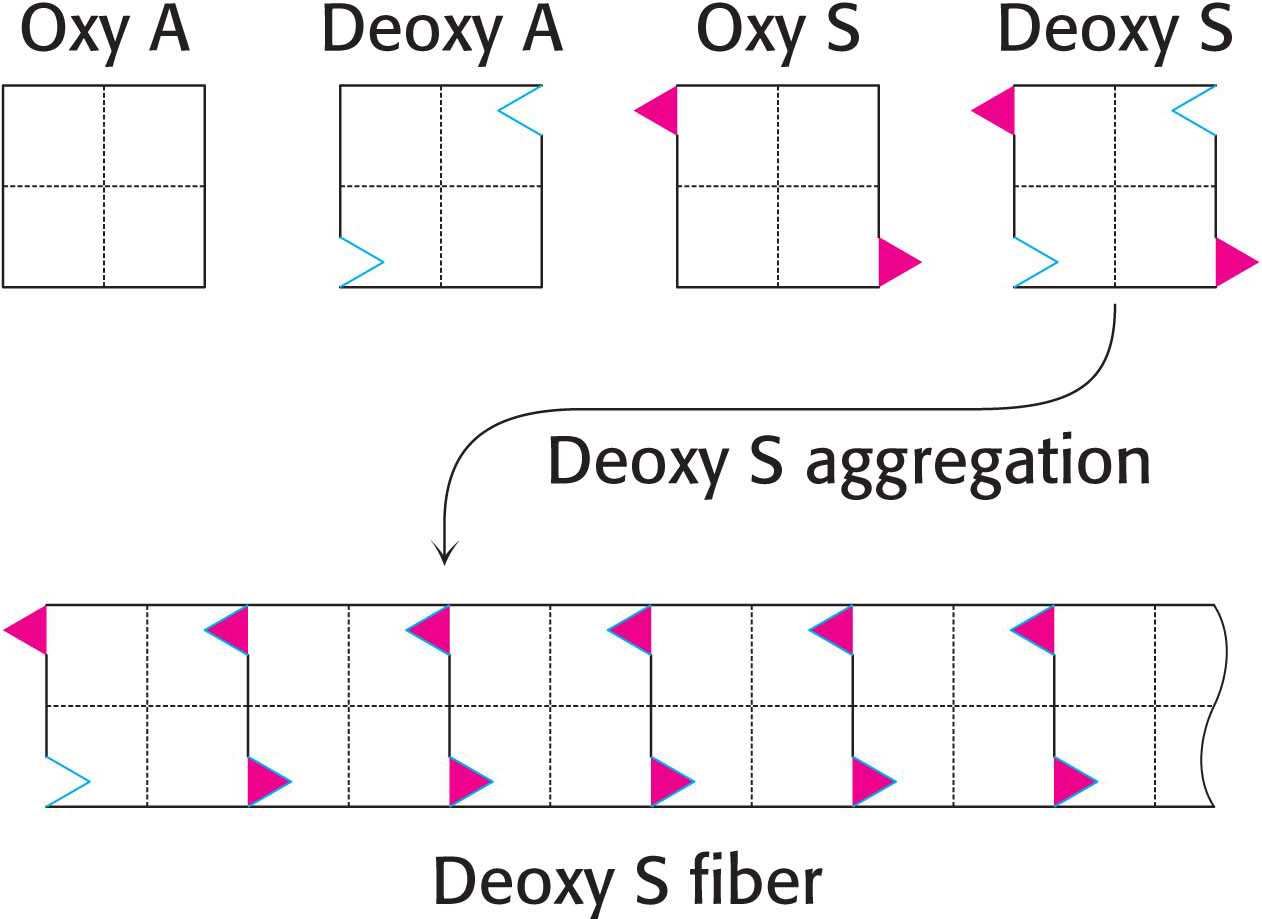
A vicious cycle is set up when sickling takes place in a small blood vessel. The blockage of the vessel creates a local region of low oxygen concentration. Hence, more hemoglobin changes into the deoxy form and so more sickling takes place. Sickled red cells become trapped in the small blood vessels, which impairs circulation and leads to the damage of multiple organs. Sickled cells, which are more fragile than normal red blood cells, rupture (hemolyze) readily to produce severe anemia. Unfortunately, effective treatment of sickle-
Approximately 1 in 100 West Africans suffer from sickle-
CLINICAL INSIGHTThalassemia is caused by an imbalanced production of hemoglobin chains
Sickle-
In β-thalassemia, the β chain of hemoglobin is not produced in sufficient quantity. In the absence of β chains, the α chains form insoluble aggregates that precipitate inside immature red blood cells. The loss of red blood cells results in anemia. The severity of thalassemia depends on how much the gene is disrupted. The most severe form of β-thalassemia, called thalassemia major or Cooley anemia, results when genes from both parents are defective. Blood transfusions are the most common treatment for thalassemias.