PROBLEMS
PROBLEMS
Question 6.1
What’s the score? Using the identity-
WYLGKITRMDAEVLLKKPTVRDGHFLVTQCESSPGEF-
WYFGKITRRESERLLLNPENPRGTFLVRESETTKGAY-
SISVRFGDSVQ-
----HFKVLRDQNGKYYLWAVK- FN- CLSVSDFDNAKGLNVKHYKIRKLDSGGFYITSRTQFS-
SLNELVAYHRTASVSRTHTILLSDMNV
SSLQQLVAYYSKHADGLCHRLTNV
Question 6.2
Sequence and structure. A comparison of the aligned amino acid sequences of two proteins each consisting of 150 amino acids reveals them to be only 8% identical. However, their three-
Question 6.3
It depends on how you count. Consider the following two sequence alignments:
A-
SNLFDIRLIG
GSNDFYEVKIMDASNLFDIRLI-
G
GSNDFYEVKIMD
189
Which alignment has a higher score if the identity-
Question 6.4
Discovering a new base pair. Examine the ribosomal RNA sequences in Figure 6.20. In sequences that do not contain Watson–
Question 6.5
Overwhelmed by numbers. Suppose that you wish to synthesize a pool of RNA molecules that contain all four bases at each of 40 positions. How much RNA must you have in grams if the pool is to have at least a single molecule of each sequence? The average molecular weight of a nucleotide is 330 g mol−1.
Question 6.6
Form follows function. The three-
Question 6.7
Shuffling. Using the identity-
ASNFLDKAGK
ATDYLEKAGK
Generate a shuffled version of sequence 2 by randomly reordering these 10 amino acids. Align your shuffled sequence with sequence 1 without allowing gaps, and calculate the alignment score between sequence 1 and your shuffled sequence.
Question 6.8
Interpreting the score. Suppose that the sequences of two proteins each consisting of 200 amino acids are aligned and that the percentage of identical residues has been calculated. How would you interpret each of the following results in regard to the possible divergence of the two proteins from a common ancestor?
80%
50%
20%
10%.
Question 6.9
Particularly unique. Consider the Blosum-
Question 6.10
A set of three. The sequences of three proteins (A, B, and C) are compared with one another, yielding the following levels of identity:
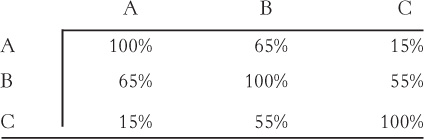
Assume that the sequence matches are distributed uniformly along each aligned sequence pair. Would you expect protein A and protein C to have similar three-
Question 6.11
RNA alignment. Sequences of an RNA fragment from five species have been determined and aligned. Propose a likely secondary structure for these fragments.
UUGGAGAUUCGGUAGAAUCUCCC
GCCGGGAAUCGACAGAUUCCCCG
CCCAAGUCCCGGCAGGGACUUAC
CUCACCUGCCGAUAGGCAGGUCA
AAUACCACCCGGUAGGGUGGUUC
Question 6.12
The more the merrier. When RNA alignments are used to determine secondary structure, it is advantageous to have many sequences representing a wide variety of species. Why?
Question 6.13
To err is human. You have discovered a mutant form of a thermostable DNA polymerase with significantly reduced fidelity in adding the appropriate nucleotide to the growing DNA strand, compared with wild-
Question 6.14
Generation to generation. When performing a molecular-
Question 6.15
BLAST away. Using the National Center for Biotechnology Information Web site (www.ncbi.nlm.nih.gov), find the sequence of the enzyme triose phosphate isomerase from E. coli strain K-
190