10.4Many Enzymes are Activated by Specific Proteolytic Cleavage
Many Enzymes are Activated by Specific Proteolytic Cleavage
We turn now to a different mechanism of enzyme regulation. Many enzymes acquire full enzymatic activity as they spontaneously fold into their characteristic three-
Specific proteolysis is a common means of activating enzymes and other proteins in biological systems. For example:
The digestive enzymes that hydrolyze foodstuffs are synthesized as zymogens in the stomach and pancreas (Table 10.3).
Blood clotting is mediated by a cascade of proteolytic activations that ensures a rapid and amplified response to trauma.
Some protein hormones are synthesized as inactive precursors. For example, insulin is derived from proinsulin by proteolytic removal of a peptide.
The fibrous protein collagen, the major constituent of skin and bone, is derived from procollagen, a soluble precursor.
Many developmental processes are controlled by the activation of zymogens. For example, in the metamorphosis of a tadpole into a frog, large amounts of collagen are resorbed from the tail in the course of a few days. Likewise, much collagen is broken down in a mammalian uterus after delivery. The conversion of procollagenase into collagenase, the active protease responsible for collagen breakdown, is precisely timed in these remodeling processes.
Programmed cell death, or apoptosis, is mediated by proteolytic enzymes called caspases, which are synthesized in precursor form as procaspases. When activated by various signals, caspases function to cause cell death in most organisms, ranging from C. elegans to human beings. Apoptosis provides a means of sculpting the shapes of body parts in the course of development and a means of eliminating damaged or infected cells.
|
Site of synthesis |
Zymogen |
Active enzyme |
|---|---|---|
|
Stomach |
Pepsinogen |
Pepsin |
|
Pancreas |
Chymotrypsinogen |
Chymotrypsin |
|
Pancreas |
Trypsinogen |
Trypsin |
|
Pancreas |
Procarboxypeptidase |
Carboxypeptidase |
We next examine the activation and control of zymogens, using enzymes responsible for digestion and blood-
Chymotrypsinogen is activated by specific cleavage of a single peptide bond
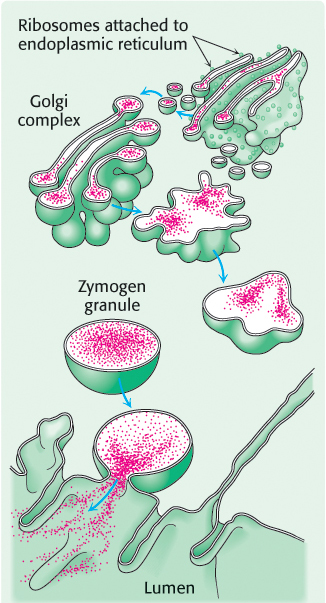
Chymotrypsin is a digestive enzyme that hydrolyzes proteins. Chymotrypsin, whose mechanism of action was described in detail in Chapter 9, specifically cleaves peptide bonds on the carboxyl side of amino acid residues with large, hydrophobic R groups (Table 8.6). Its inactive precursor, chymotrypsinogen, is synthesized in the pancreas, as are several other zymogens and digestive enzymes. Indeed, the pancreas is one of the most active organs in synthesizing and secreting proteins. The enzymes and zymogens are synthesized in the acinar cells of the pancreas and stored inside membrane-
300
Chymotrypsinogen, a single polypeptide chain consisting of 245 amino acid residues, is virtually devoid of enzymatic activity. It is converted into a fully active enzyme when the peptide bond joining arginine 15 and isoleucine 16 is cleaved by trypsin (Figure 10.20). The resulting active enzyme, called π-chymotrypsin, then acts on other π-chymotrypsin molecules by removing two dipeptides to yield α-chymotrypsin, the stable form of the enzyme. The three resulting chains in α-chymotrypsin remain linked to one another by two interchain disulfide bonds. The striking feature of this activation process is that cleavage of a single specific peptide bond transforms the protein from a catalytically inactive form into one that is fully active.
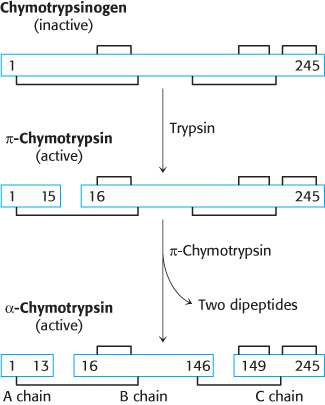
Proteolytic activation of chymotrypsinogen leads to the formation of a substrate-binding site
How does cleavage of a single peptide bond activate the zymogen? The cleavage of the peptide bond between amino acids 15 and 16 triggers key conformational changes, which were revealed by the elucidation of the three-
The newly formed amino-
terminal group of isoleucine 16 turns inward and forms an ionic bond with aspartate 194 in the interior of the chymotrypsin molecule (Figure 10.21).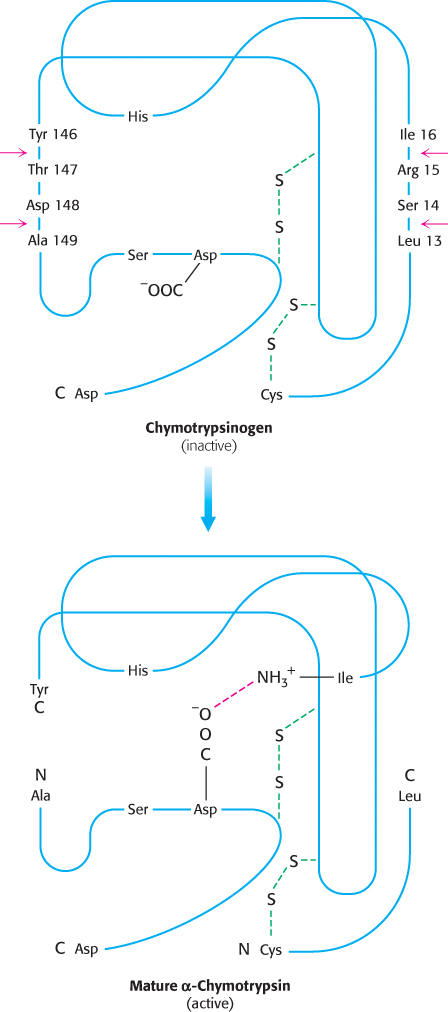 FIGURE 10.21Conformations of chymotrypsinogen and chymotrypsin. The electrostatic interaction between the α-amino group of isoleucine 16 and the carboxylate of aspartate 194, essential for the structure of active chymotrypsin, is possible only only after cleavage of the peptide bond between isoleucine and arginine in chymotrypsinogen.[Information from Gregory A. Petsko and Dagmar Ringe, Protein Structure and Function (Sinauer, 2003), p. 3-
FIGURE 10.21Conformations of chymotrypsinogen and chymotrypsin. The electrostatic interaction between the α-amino group of isoleucine 16 and the carboxylate of aspartate 194, essential for the structure of active chymotrypsin, is possible only only after cleavage of the peptide bond between isoleucine and arginine in chymotrypsinogen.[Information from Gregory A. Petsko and Dagmar Ringe, Protein Structure and Function (Sinauer, 2003), p. 3-16, Figure 3- 31.] This electrostatic interaction triggers a number of conformational changes. Methionine 192 moves from a deeply buried position in the zymogen to the surface of the active enzyme, and residues 187 and 193 move farther apart from each other. These changes result in the formation of the substrate-
specificity site for aromatic and bulky nonpolar groups. One side of this site is made up of residues 189 through 192. This cavity for binding part of the substrate is not fully formed in the zymogen.The tetrahedral transition state generated by chymotrypsin has an oxyanion (a negatively charged carbonyl oxygen atom) that is stabilized by hydrogen bonds with two NH groups of the main chain of the enzyme (Figure 9.9). One of these NH groups is not appropriately located in chymotrypsinogen, and so the site stabilizing the oxyanion (the oxyanion hole) is incomplete in the zymogen.
The conformational changes elsewhere in the molecule are very small. Thus, the switching on of enzymatic activity in a protein can be accomplished by discrete, highly localized conformational changes that are triggered by the hydrolysis of a single peptide bond.
301
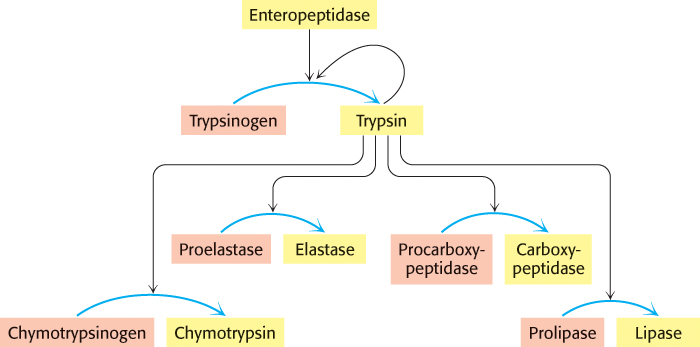
The generation of trypsin from trypsinogen leads to the activation of other zymogens
The structural changes accompanying the activation of trypsinogen, the precursor of the proteolytic enzyme trypsin, are different from those in the activation of chymotrypsinogen. Four regions of the polypeptide are very flexible in the zymogen, whereas they have a well-
The digestion of proteins and other molecules in the duodenum requires the concurrent action of several enzymes, because each is specific for a limited number of side chains. Thus, the zymogens must be switched on at the same time. Coordinated control is achieved by the action of trypsin as the common activator of all the pancreatic zymogens —trypsinogen, chymotrypsinogen, proelastase, procarboxypeptidase, and prolipase, the inactive precursor of a lipid-
302
Some proteolytic enzymes have specific inhibitors
The conversion of a zymogen into a protease by cleavage of a single peptide bond is a precise means of switching on enzymatic activity. However, this activation step is irreversible, and so a different mechanism is needed to terminate proteolysis. Specific protease inhibitors accomplish this task. Serpins, serine protease inhibitors, are an example of one such family of inhibitors. For example, pancreatic trypsin inhibitor, a 6-
The reason for the exceptional stability of the complex is that pancreatic trypsin inhibitor is a very effective substrate analog. X-
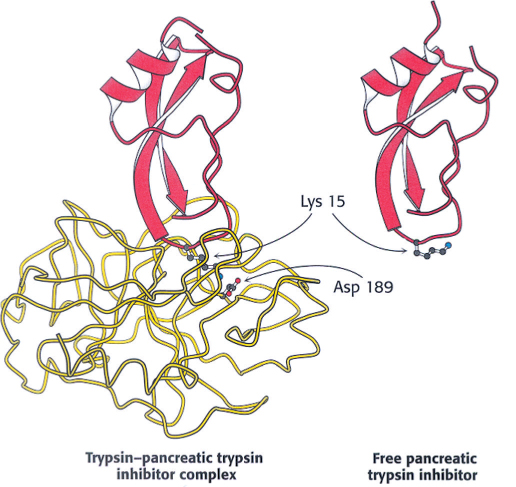
 The amount of trypsin is much greater than the amount of inhibitor. Why does trypsin inhibitor exist? Recall that trypsin activates other zymogens. Consequently, the prevention of even small amounts of trypsin from initiating the cascade while the zymogens are still in the pancreas or pancreatic ducts is vital. Trypsin inhibitor binds to any prematurely activated trypsin molecules in the pancreas or pancreatic ducts. This inhibition prevents severe damage to those tissues, which could lead to acute pancreatitis.
The amount of trypsin is much greater than the amount of inhibitor. Why does trypsin inhibitor exist? Recall that trypsin activates other zymogens. Consequently, the prevention of even small amounts of trypsin from initiating the cascade while the zymogens are still in the pancreas or pancreatic ducts is vital. Trypsin inhibitor binds to any prematurely activated trypsin molecules in the pancreas or pancreatic ducts. This inhibition prevents severe damage to those tissues, which could lead to acute pancreatitis.
303
Pancreatic trypsin inhibitor is not the only important protease inhibitor. A 53-
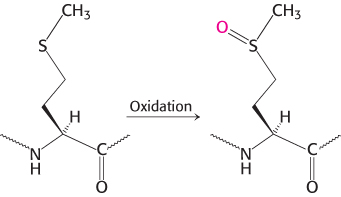
The resulting clinical condition is called emphysema (also known as chronic obstructive pulmonary disease [COPD]). People with emphysema must breathe much harder than normal people to exchange the same volume of air because their alveoli are much less resilient than normal. Cigarette smoking markedly increases the likelihood that even a type Z heterozygote will develop emphysema. The reason is that smoke oxidizes methionine 358 of the inhibitor (Figure 10.24), a residue essential for binding elastase. Indeed, this methionine side chain is the bait that selectively traps elastase. The methionine sulfoxide oxidation product, in contrast, does not lure elastase, a striking consequence of the insertion of just one oxygen atom into a protein and a remarkable example of the effect of human behavior on biochemistry. We will consider another protease inhibitor, antithrombin III, when we examine the control of blood clotting.
Blood clotting is accomplished by a cascade of zymogen activations
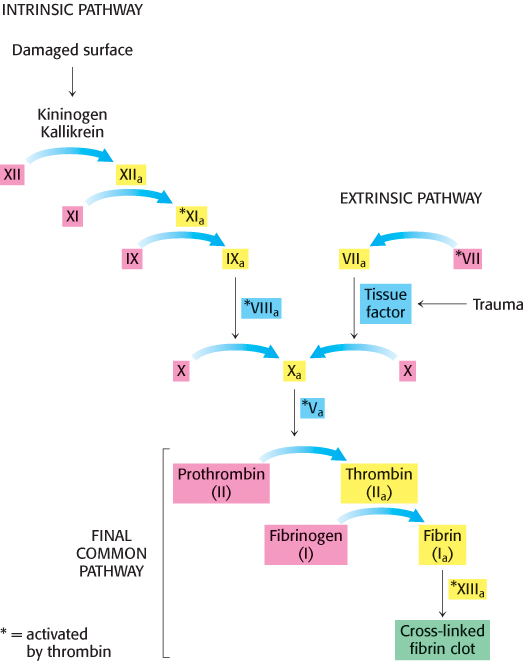
Enzymatic cascades are often employed in biochemical systems to achieve a rapid response. In a cascade, an initial signal institutes a series of steps, each of which is catalyzed by an enzyme. At each step, the signal is amplified. For instance, if a signal molecule activates an enzyme that in turn activates 10 enzymes and each of the 10 enzymes in turn activates 10 additional enzymes, after four steps the original signal will have been amplified 10,000-
304
Two means of initiating blood clotting have been described, the intrinsic pathway and the extrinsic pathway. The intrinsic clotting pathway is activated by exposure of anionic surfaces upon rupture of the endothelial lining of the blood vessels. The extrinsic pathway, which appears to be most crucial in blood clotting, is initiated when trauma exposes tissue factor (TF), an integral membrane glycoprotein. Upon exposure to the blood, tissue factor binds to factor VII to activate factor X. Both the intrinsic and extrinsic pathways lead to the activation of factor X (a serine protease), which in turn converts prothrombin into thrombin, the key protease in clotting. Thrombin then amplifies the clotting process by activating enzymes and factors that lead to the generation of yet more thrombin, an example of positive feedback. Note that the active forms of the clotting factors are designated with a subscript “a,” whereas factors that are activated by thrombin are designated with an asterisk.

Prothrombin requires a vitamin K-dependent modification for activation
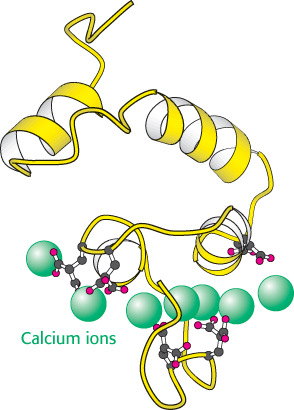
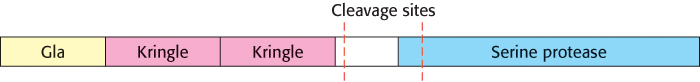
Thrombin is synthesized as a zymogen called prothrombin. The inactive molecule comprises four major domains, with the serine protease domain at its carboxyl terminus (Figure 10.26). The first domain, called the gla domain, is rich in γ carboxyglutamate residues (abbreviation gla), and the second and third domains are called kringle domains (named after a Danish pastry that they resemble). Vitamin K is required for the synthesis of γ carboxyglutamate, a strong chelator of Ca2+. These three domains work in concert to keep prothrombin in an inactive form. Moreover, because it is rich in γ carboxyglutamate, the gla domain is able to bind Ca2+ (Figure 10.27). What is the effect of this binding? The binding of Ca2+ by prothrombin anchors the zymogen to phospholipid membranes derived from blood platelets after injury. This binding is crucial because it brings prothrombin into close proximity to two clotting proteins, factor Xa and factor Va (a stimulatory protein), that catalyze its conversion into thrombin. Activation is begun by proteolytic cleavage of the bond between arginine 274 and threonine 275 to release a fragment containing the first three domains. Cleavage of the bond between arginine 323 and isoleucine 324 yields active thrombin.
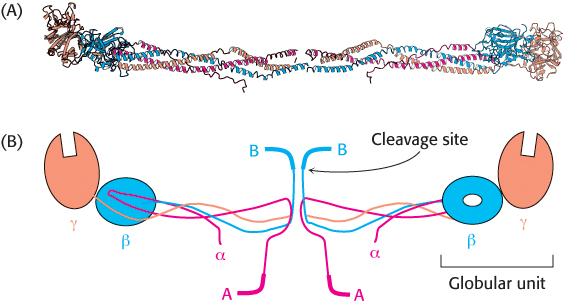
Fibrinogen is converted by thrombin into a fibrin clot
The best-
305
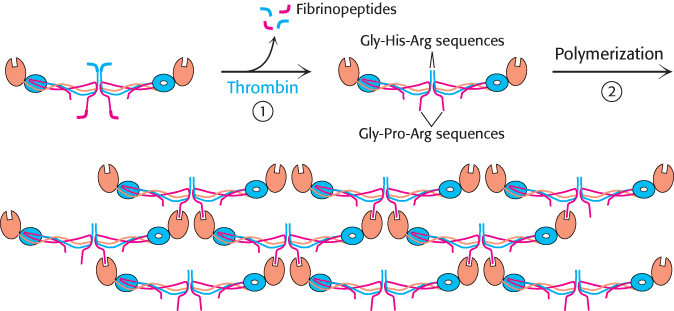
Fibrin monomers spontaneously assemble into ordered fibrous arrays called fibrin. Electron micrographs and low-

306
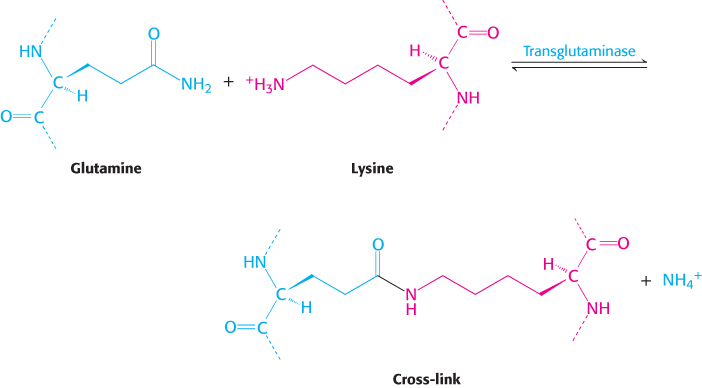
This cross-
Vitamin K is required for the formation of γ-carboxyglutamate
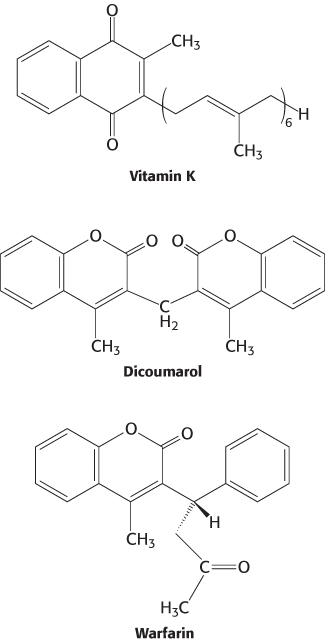
Vitamin K (Figure 10.31) has been known for many years to be essential for the synthesis of prothrombin and several other clotting factors. Indeed, it is called vitamin K because a deficiency in this vitamin results in defective blood koagulation (Scandinavian spelling). After ingestion, vitamin K is reduced to a dihydro derivative that is required by γ-glutamyl carboxylase to convert the first 10 glutamate residues in the amino-
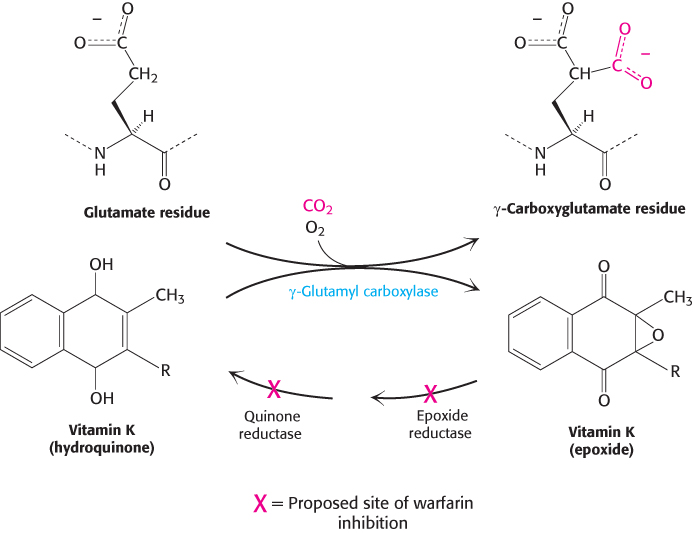
307
An account of a hemorrhagic disposition existing in certain families
“About seventy or eighty years ago, a woman by the name of Smith settled in the vicinity of Plymouth, New Hampshire, and transmitted the following idiosyncrasy to her descendants. It is one, she observed, to which her family is unfortunately subject and has been the source not only of great solicitude, but frequently the cause of death. If the least scratch is made on the skin of some of them, as mortal a hemorrhage will eventually ensue as if the largest wound is inflicted…. It is a surprising circumstance that the males only are subject to this strange affection, and that all of them are not liable to it…. Although the females are exempt, they are still capable of transmitting it to their male children.”
John Otto (1803)
Recall that γcarboxyglutamate, a strong chelator of Ca2+, is required for the activation of prothrombin. Dicoumarol, which is found in spoiled sweet clover, causes a fatal hemorrhagic disease in cattle fed on this hay. Cows fed dicoumarol synthesize an abnormal prothrombin that does not bind Ca2+, in contrast with normal prothrombin. Dicoumarol was the first anticoagulant used to prevent thromboses in patients prone to clot formation. However, it is seldom used now because of poor absorption and gastrointestinal side effects. Warfarin, another vitamin K antagonist, is commonly administered as an anticoagulant. Warfarin inhibits the keto reductase and quinone reductase that are required to regenerate the dihydro derivative of vitamin K (Figure 10.32). Dicoumarol, warfarin, and their chemical derivatives serve as effective rat poisons.
The clotting process must be precisely regulated
There is a fine line between hemorrhage and thrombosis, the formation of blood clots in blood vessels. Clots must form rapidly yet remain confined to the area of injury. What are the mechanisms that normally limit clot formation to the site of injury? The lability of clotting factors contributes significantly to the control of clotting. Activated factors are short-
Specific inhibitors of clotting factors are also critical in the termination of clotting. For instance, tissue factor pathway inhibitor (TFPI) inhibits the complex of TF–
The importance of the ratio of thrombin to antithrombin is illustrated in the case of a 14-
Antithrombin limits the extent of clot formation, but what happens to the clots themselves? Clots are not permanent structures but are designed to dissolve when the structural integrity of damaged areas is restored. Fibrin is degraded by plasmin, a serine protease that hydrolyzes peptide bonds in the coiled-

308
Hemophilia revealed an early step in clotting
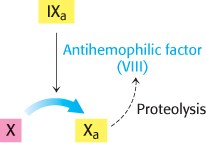
Some important breakthroughs in the elucidation of clotting pathways have come from studies of patients with bleeding disorders. Classic hemophilia, or hemophilia A, is the best-
In the past, hemophiliacs were treated with transfusions of a concentrated plasma fraction containing factor VIII. This therapy carried the risk of infection. Indeed, many hemophiliacs contracted hepatitis and, more recently, AIDS. A safer source of factor VIII was urgently needed. With the use of biochemical purification and recombinant DNA techniques, the gene for factor VIII was isolated and expressed in cells grown in culture. Recombinant factor VIII purified from these cells has largely replaced plasma concentrates in treating hemophilia.